M. Cardoso-Moreia et al.: Gene expression across mammalian organ development. Nature 571:505 (2019).
Spezifisch arbeiten wir mit der Expressions-Matrix, die aus den Maus-Proben gebildet wurde. Zusammen mit den Rohdaten haben die Autoren diese Matrix als Datei Mouse.CPM.txt verfügbar gemacht. Allerdings wurden in dieser Datei die rohen Read-Zahlen bereits in CPM-Werte umgewandelt. Da wir am Anfang beginnen wollen, habe ich diese Normalisierung rückgängig gemacht (siehe hier für Details); die Matrix, die ich so erhalten habe, ist hier hier verfügbar).
Rows: 36141 Columns: 317
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): gene_id
dbl (316): Brain_e10.5_1, Brain_e10.5_2, Brain_e10.5_3, Brain_e11.5_1, Brain...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Hier sind die ersten Spalten und Zeilen der Matrix:
Die Tabelle enthält Daten von 316 Proben. Wir verschaffen uns zunächst einen Überblick über diese, indem wir die Spaltennamen interpretieren. Jeder Spaltennamen besteht aus drei Teilen, die durch Unterstriche getrennt sind: dem Gewebe (bei der ersten Spalte: Brain), dem Zeitpunkt (e10.5) und der Nummer des Replikats (d.h., des Embryos oder Tieres; hier 1).
Wir erzeugen eine Tabelle mit den Spaltennamen, die wir als Probennamen (“sample names”) verwenden werden. Den Namen der ersten Spalte verwenden wir nicht, da das die Gen-Namen sind.
Hier haben wir timepoint zu einem Faktor gemacht, um sicherzustellen, dass die Reihenfolge der Zeitpunkte später beim Plotten nicht verändert wird. Wir haben nun folgende Zeitpunkte:
Hier bedeutet “e10.5”, dass die Probe von einem Embryo stammt, 10.5 Tage nach der Befruchtung, und “P3”, dass sie von einem Tier stammt, 3 Tage nach der Geburt. (Die Funktion levels gibt die verschiedenen Werte eine Faktors in der festgelegten Reihenfolge aus.)
Erkennen Sie, wie der Code funktioniert? Sehen Sie sich dazu an, wie die Ausgabe aussieht ohne die letzte Zeile. // Für Fortgeschrittene: Was macht wohl das values_fill? Was geschieht, wenn man es weglässt, und warum geschieht das?
Hochexprimierte Gene; Gen-Namen
Nun sollten wir einen ersten Blick auf unsere Count-Matrix werfen:
Welche Gene sind wohl in der Leber besonders hoch exprimiert? Wir nehmen uns eine Probe heraus, z.B. die erste Probe mit Leber neugeborener Mäuse, also Liver_P0_1 und sortieren die Tabelle absteigend (“descending”)
Mit Google lässt sich schnell ermittlen, dass die Ensembl-ID ENSMUSG00000029368, die nun ganz oben steht, für das Gen Alb steht, das für Albumin kodiert, das Protein, das im Blutserum mit der höchsten Konzentration vorkommt. Dass die Leberzellen sehr viel mRNA für dieses Gen brauchen, um dieses wichtige Protein in der erfordeelichen großen Menge zu erzeugen, leuchtet ein.
Was sind die anderen Gene? Wir laden die Datei mit der Zuordnung von Ensembl-Gen-IDs zu Gen-Symbolen , die wir bereits das letzte Mal erstellt haben. Wir benennen aber die Spalten um, um sie etwas kürzer zu machen:
Rows: 56305 Columns: 3
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (3): Gene stable ID, Gene name, Gene description
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Auch die anderen hochexprimierten Gene passen zur Aufgaben der Leber.
Werfen wir zum Vergleich noch einen Blick auf die Gene, die in der Niere am höchsten exprimiert sind. Wir verwenden wieder das erste Replikat von Zeitpunkt P0:
Dieses Mal finden wir hauptsächlich basale Transkriptionsfaktoren und ähnliches. Das klingt nicht allzu spezifisch für die Niere. Vielleicht kommen wir später darauf zurück.
Normalisierung
Zurück zu unserer großen Count-Matrix. Hier nochmal die Ecke links oben.
Das hat scheinbar nichts geändert, aber nun stehen uns spezielle Matrix-Funktionen zur Verfügung, z.B. colSum, um die Spaltensummen (“column sums”) zu berechnen
Wenn wir aber keine neuen Funktionen lernen möchten, können wir auch bei Tidyverse bleiben. Dann müssen wir aber zunächst unsere “breite” Tabelle in eine lange umwandeln, um besser damit arbeiten zu können:
Wir sehen, dass sich die Gesamtzahl der Reads recht stark von Probe zu Probe variiert. Dies hat aber keine biologische Bedeutung; es spiegelt schlicht wieder, wie gut es gelungen ist, die DNA dieser Probe effizient in die Flowcell zu füllen. Von Bedeutung ist daher nicht die absolute Anzahl der reads, die auf ein Gen gefallen sind, sondern deren Anteil an allen Reads der Probe.
Daher sollten wir jede Read-Zahl durch diese Gesamtzahl teilen. Einfach nur, um bequemere Zahlen zu haben, multiplizieren wir anschließen mit 1 Milliion (1e6). Das ergebnis nennen wir “counts per million” (“CPM”):
Hier haben wir jede Zeile der Maztrix durch ihre Spaltensumme (aus colSums) geteilt. Da aber R “matrix / vector” interpretiert als Aufforderung, jede Zeile (und nicht: Spalte) der Matrix durch ein Element des vektors zu teilen, müssen wir die Matrix vorher mit t transponieren (d.h. Zeilen und Spalten vertauschen) und nachd er Division wieder zurück transponieren.
Ob man auf diese Weise arbeitet, oder mit Tidyverse, ist Geschmackssache.
Entwicklung der Leber-Gene: Linienplots
Sehen wir uns nochmal die Liste der Gene an, die in der ersten P0-Leber-Probe am stärsten waren:
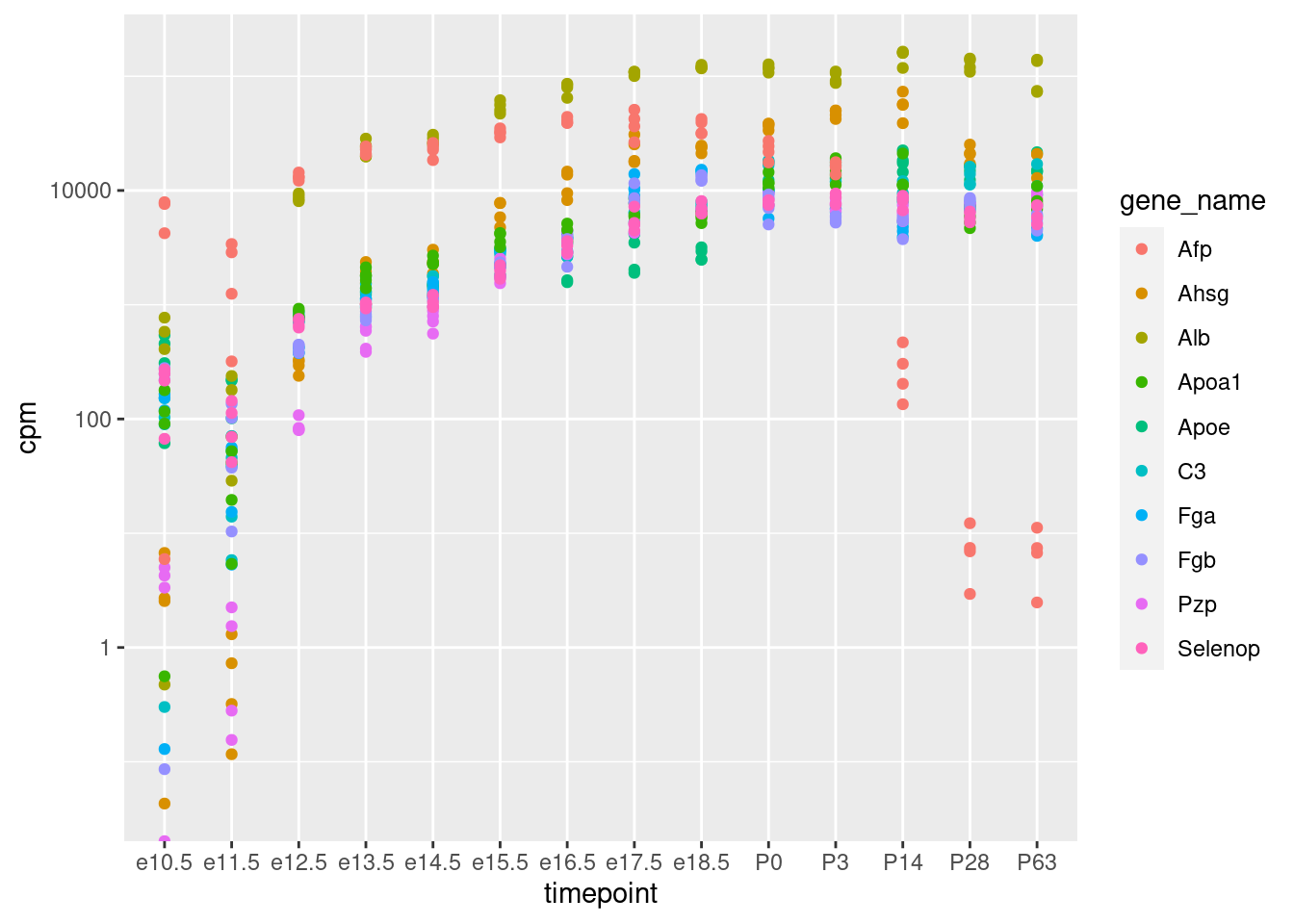
Sind diese Gene in den anderen Zeitpunkten auch so hoch? Um dies zu untersuchen, filtern wir die lange Tabelle herunter auf diese Gene, sowie auf alle Leber-Proben:
Warning: Transformation introduced infinite values in continuous y-axis
Vielleicht wäre es übersichtlicher, wenn wir Linien statt Punkten verwenden würden. Es macht aber keinen Sinn, alle Punkte, die dieselbe Replikat-Ummer haben, mit einer Linie zu verbinden, denn das sind ja dennoch verschiedene Proben. Wir versuchen, über die Replikate zu mitteln:
`summarise()` has grouped output by 'timepoint'. You can override using the
`.groups` argument.
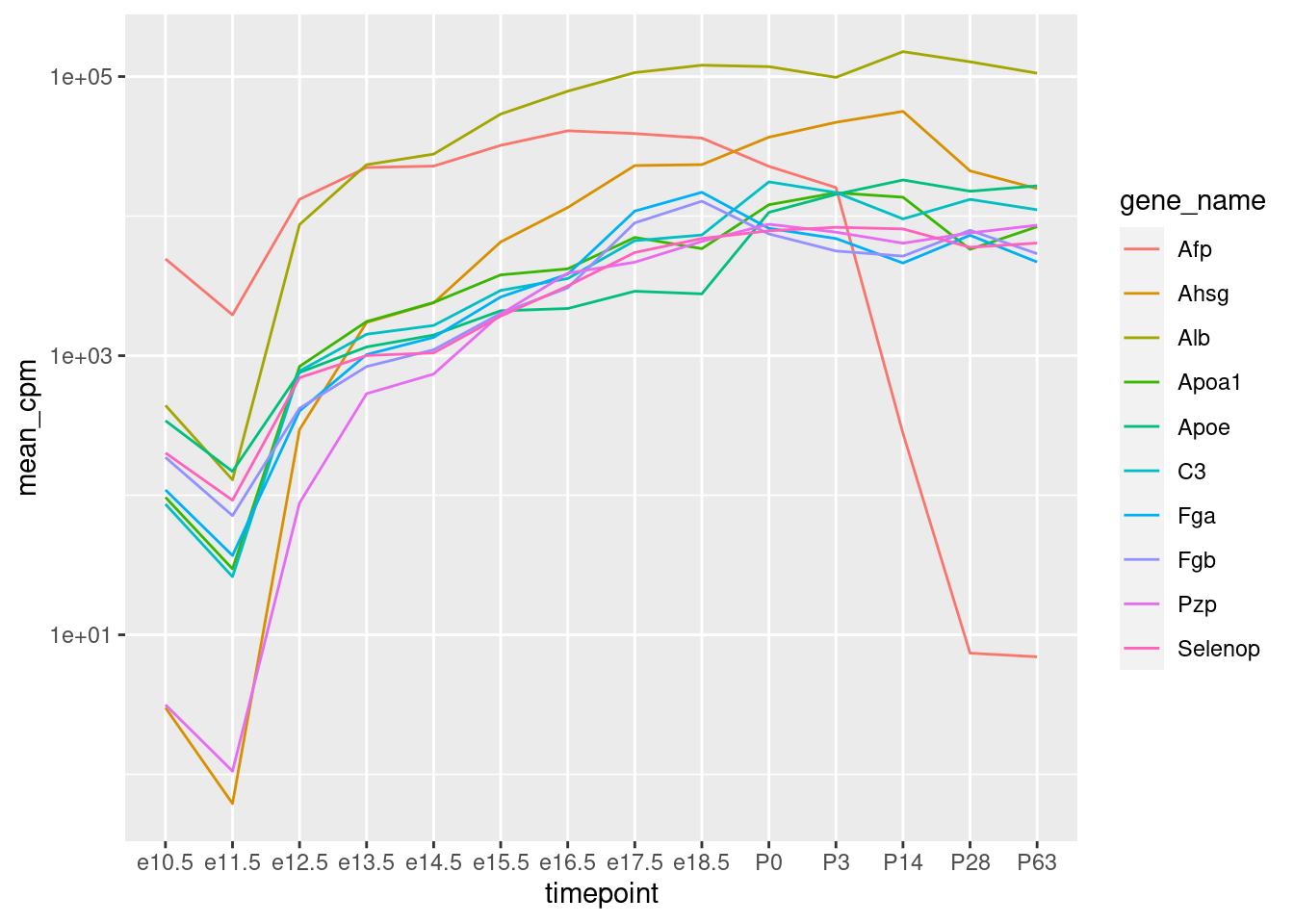
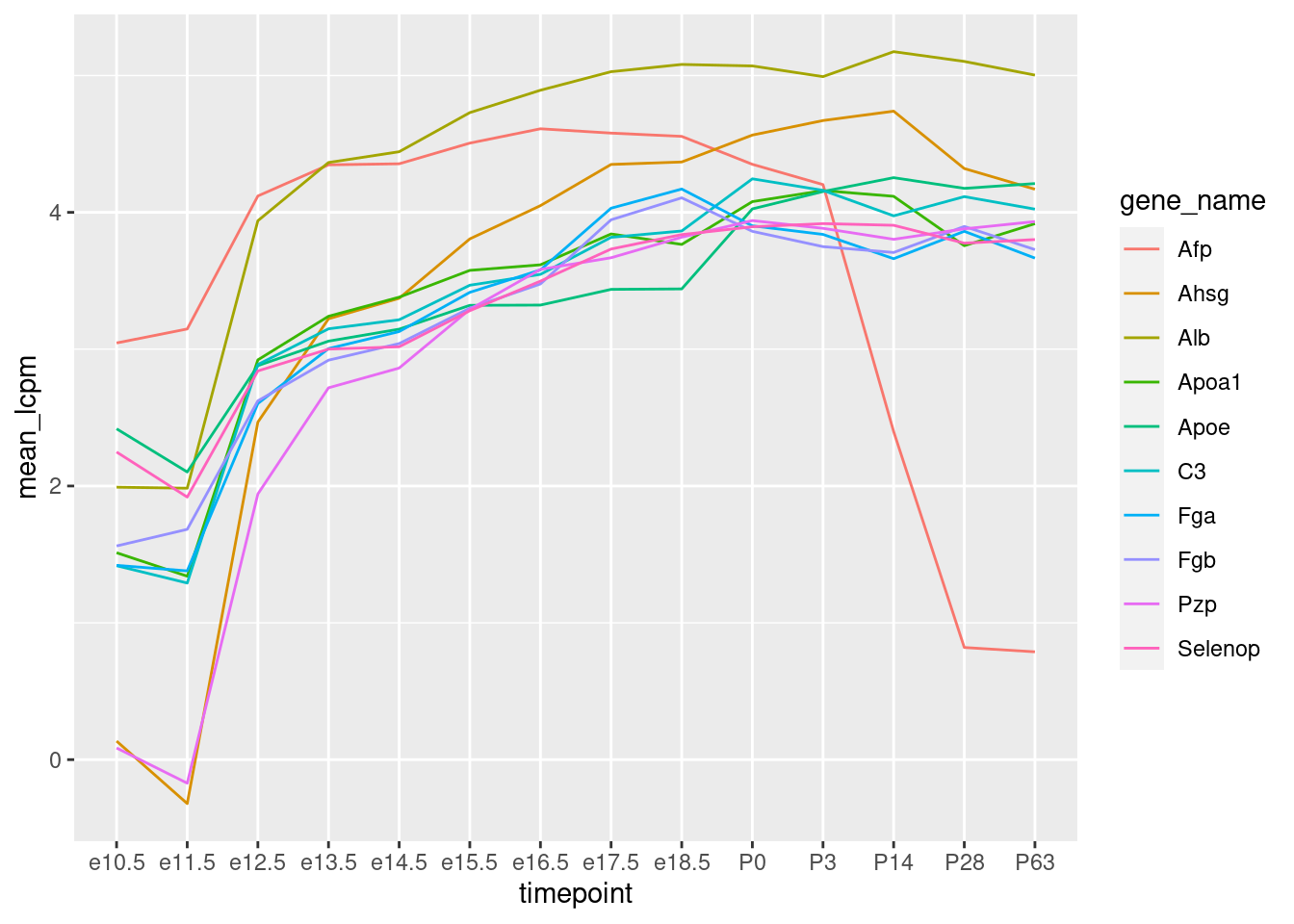
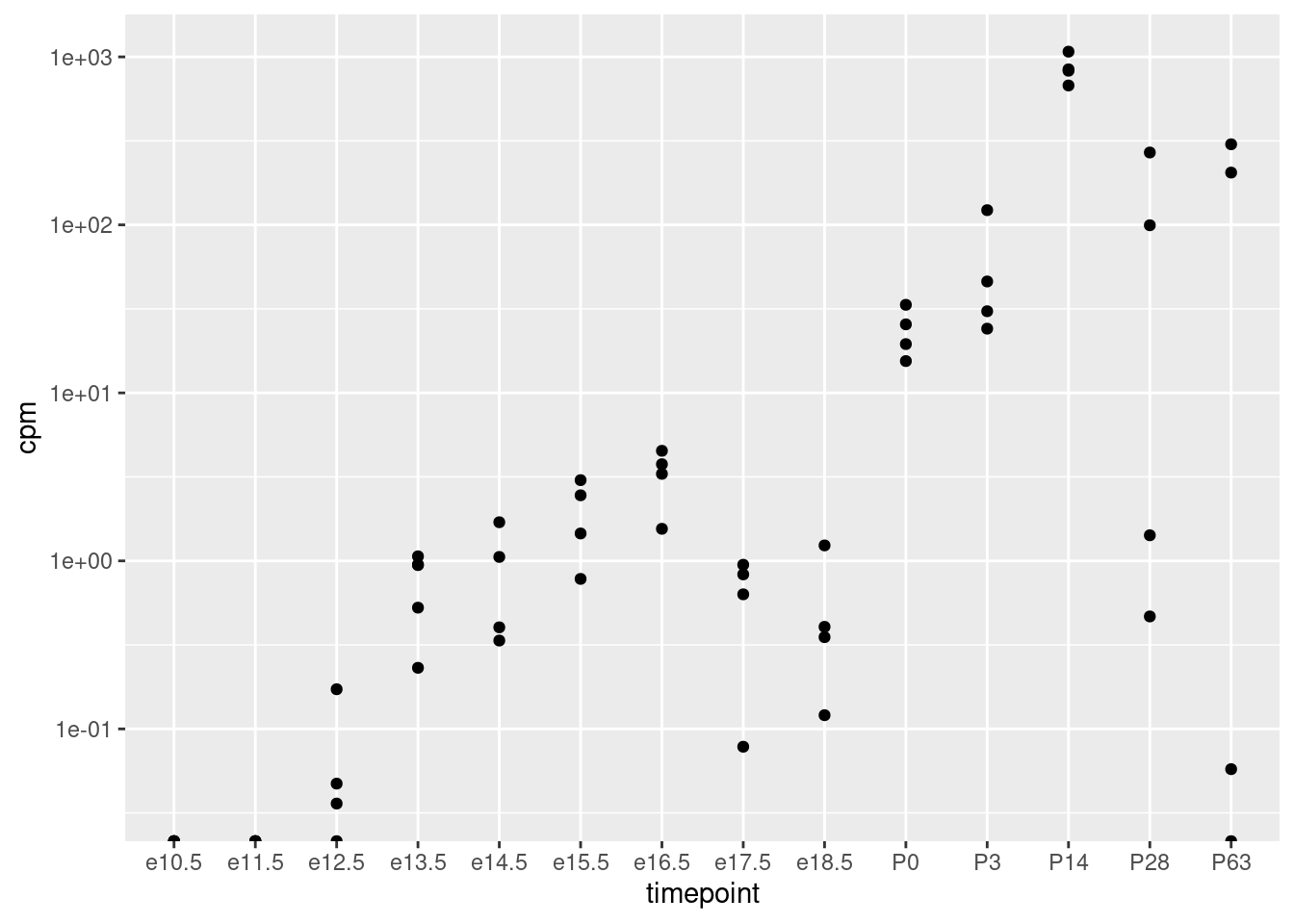
Wir erkennen, dass die meisten der 10 Gene Anfangs schwach exprimiert sind, ab E12.5 schon recht stark und dann langsam weiter zunehmen. Nach der Geburt bleiben sie in etwa konstant. Ein Gen (“Afp”) fällt wieder stark ab. “Afp” steht für “alpha-1-Fetoprotein”, und der Name zeigt schon, dass es sich um ein Protein handelt, dass wohl nur in Föten vorkommt.
Log-Expression
Wir haben eben die y-Achse logarithmiert – und erst so wird der Plot gut lesbar. Das liegt daran, dass Gen-Expressionen über viele Größenordnungen schwanken.
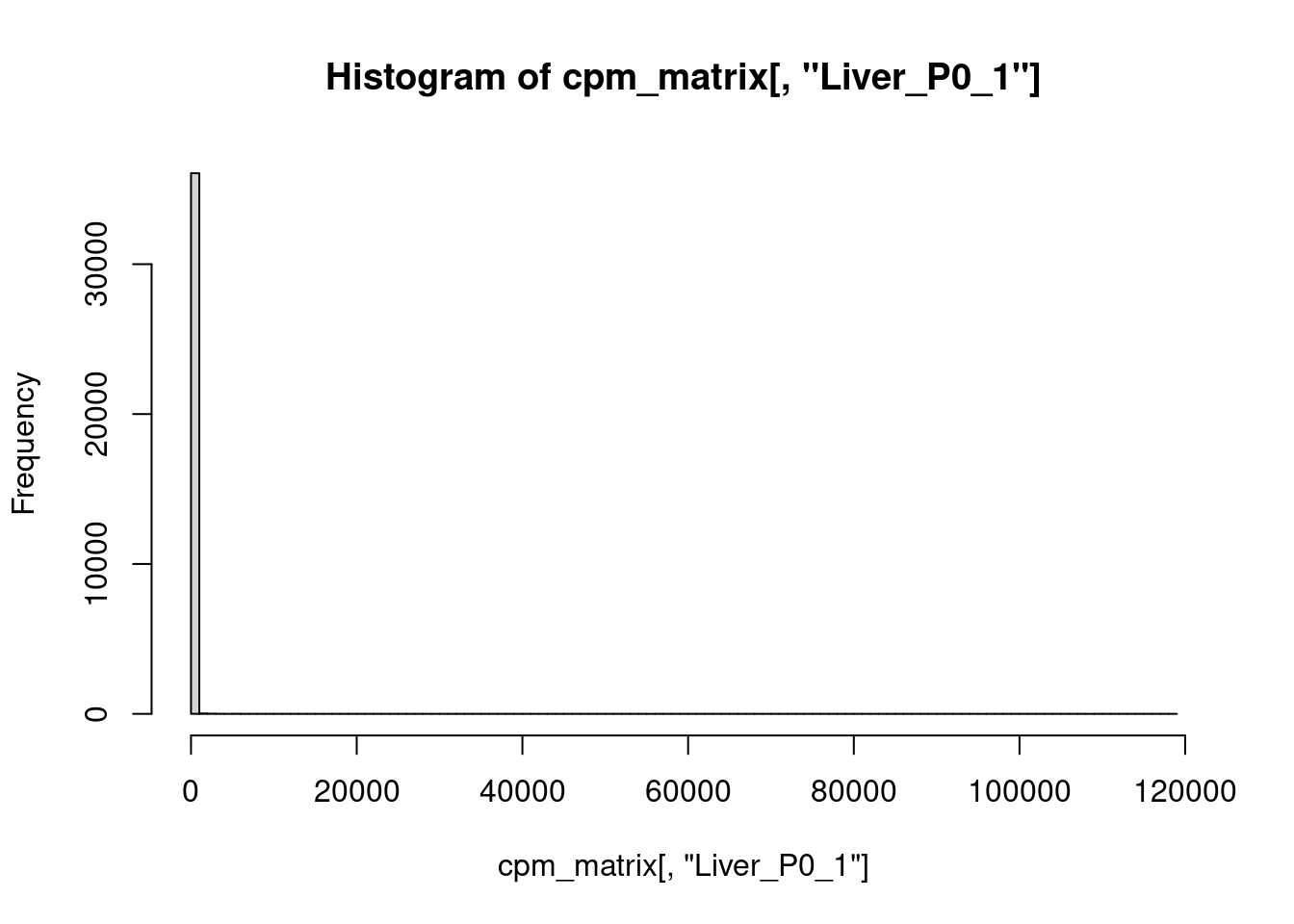
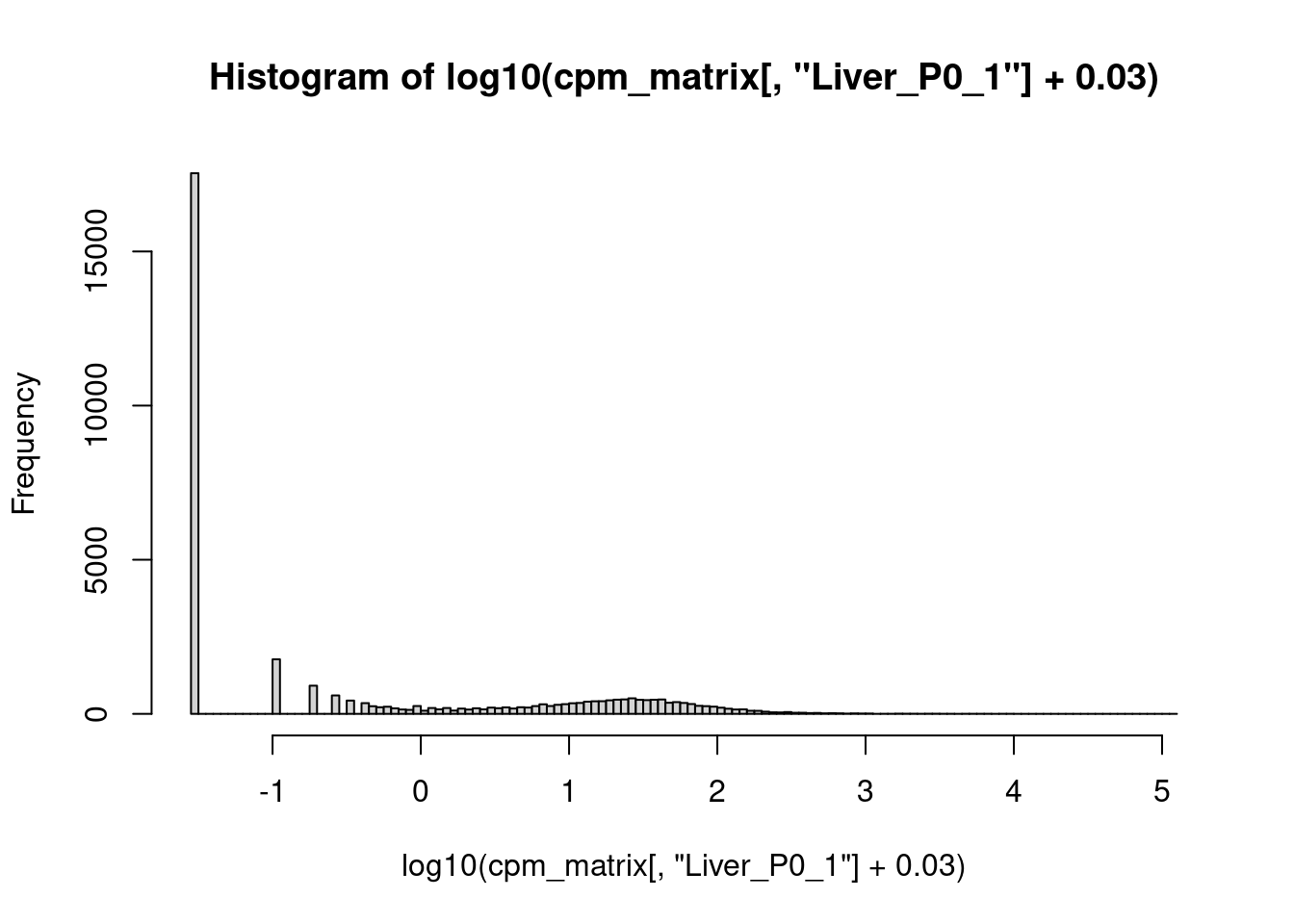
Nehmen wir dazu nochmal eine Probe heraus, wieder “Liver_P0_1”, und plotten ein Histogramm aller cpm-Werte (die wir der entsprechenden Spalte der cpm-Matrix von oben entnehmen):
hist( cpm_matrix[,"Liver_P0_1"], 100 )
Das war nicht hilfreich.
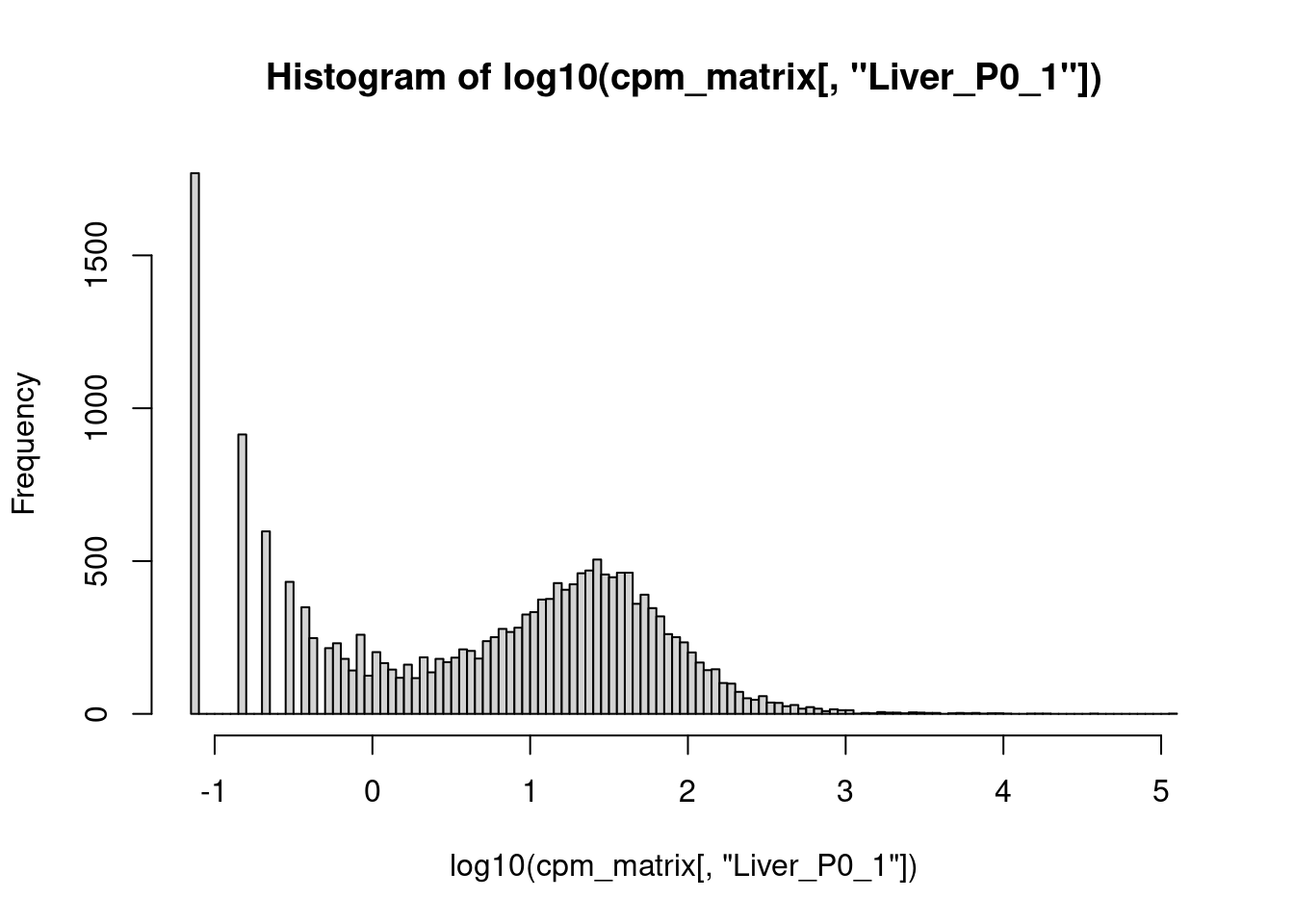
Wenn wir aber den Logarithmus ziehen, wird es besser:
hist( log10( cpm_matrix[,"Liver_P0_1"] ), 100 )
Wir sehen, dass die meisten Gene zwischen \(10^{0.5}\approx 3\) und \(10^2=100\) cpm haben, einige wenige aber über \(10^4=10000\).
Klar ist auch, dass die log10(CPM)-Werte, die von -1 bis 5 gehen, besser geeignet scheinen, um damit zu arbeiten, als die CPM-Werte, die zwar theoretisch von 0 bis über 100.000 gehen, meist aber unter 100 bleiben. Deshalb werden wir im folgenden meist mit logarithmierten Werten arbeiten.
Der Logarithmus von 0 ist minus unendlich, und daher gehen uns alle Nullen in der CPM-Matrix verloren. Um sie zu behalten, zählen wir zu jedem CPM-Wert den Wert 0.03 hinzu. Damit wird 0 zu \(\log_{10}(0.03)=-1.5\), und die anderen Werte schieben ein ganz kleines bisschen nach rechts:
Wenn wir nun auf unseren Linien-Plot oben zurück gehen, stellt sich eien Frage. Dort haben wir zwar mit der logarithmierten y-Achse die Expression bereits logarithmisch dargestellt, aber: Sollten wir erst den Logarithmus ziehen und dann über die Replikate mitteln, oder anders herum? Letzteres ist tatsächlich besser, weil sonst ein einzelnes stark exprimiertes Replikat zu viel Einfluss auf den Mittelwert hätte.
Also, hier der Linienplot nochmal, nun mit Mittelung nach dem Logarithmus.
Wir erstellen erst eine Tabelle mit allen Genen, in der wir den Logarithmus der CPM-Werte nehmen und dann über die Replikate mitteln:
Joining with `by = join_by(sample)`
`summarise()` has grouped output by 'tissue', 'timepoint'. You can override
using the `.groups` argument.
Joining with `by = join_by(gene_id)`
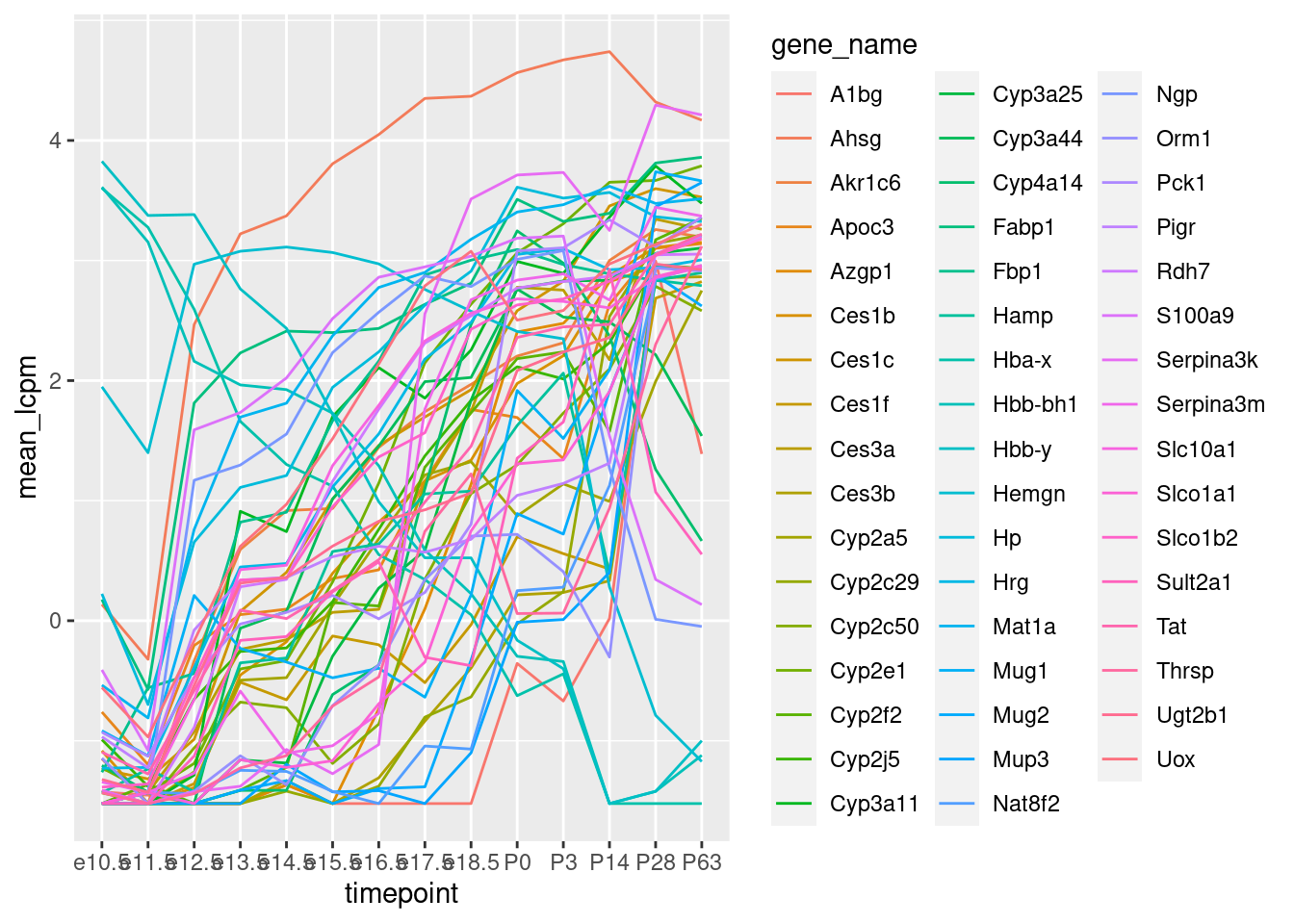
Unsere Auswahl der 10 Gene war nicht wirklich geeignet, um Gene zu finden, die sich in der Entwicklung stark ändern. Versuchen wir etwas besseres: WIr berechnen für jedes Gen den kleinsten und den größten Wert, den es über alle Zeitpunkte annimmt:
Beachten Sie: Wir haben im vorigen Schritt über die Replikate gemittelt. Nun haben wir alle diese Mittelwerte nach Genen gruppiert, d.h., die Werte vom selben Gen, aber von verschiedenen Zeitpunkten zusammen genommen, und hier die Minima und Maxima ermittelt. Dann haben wir nach der Differenz zwischen Maximum und Minimum (also der Spanne der Werte) absteigend sortiert.
Wir schreiben uns die 50 Gene mit der größten Spanne heraus:
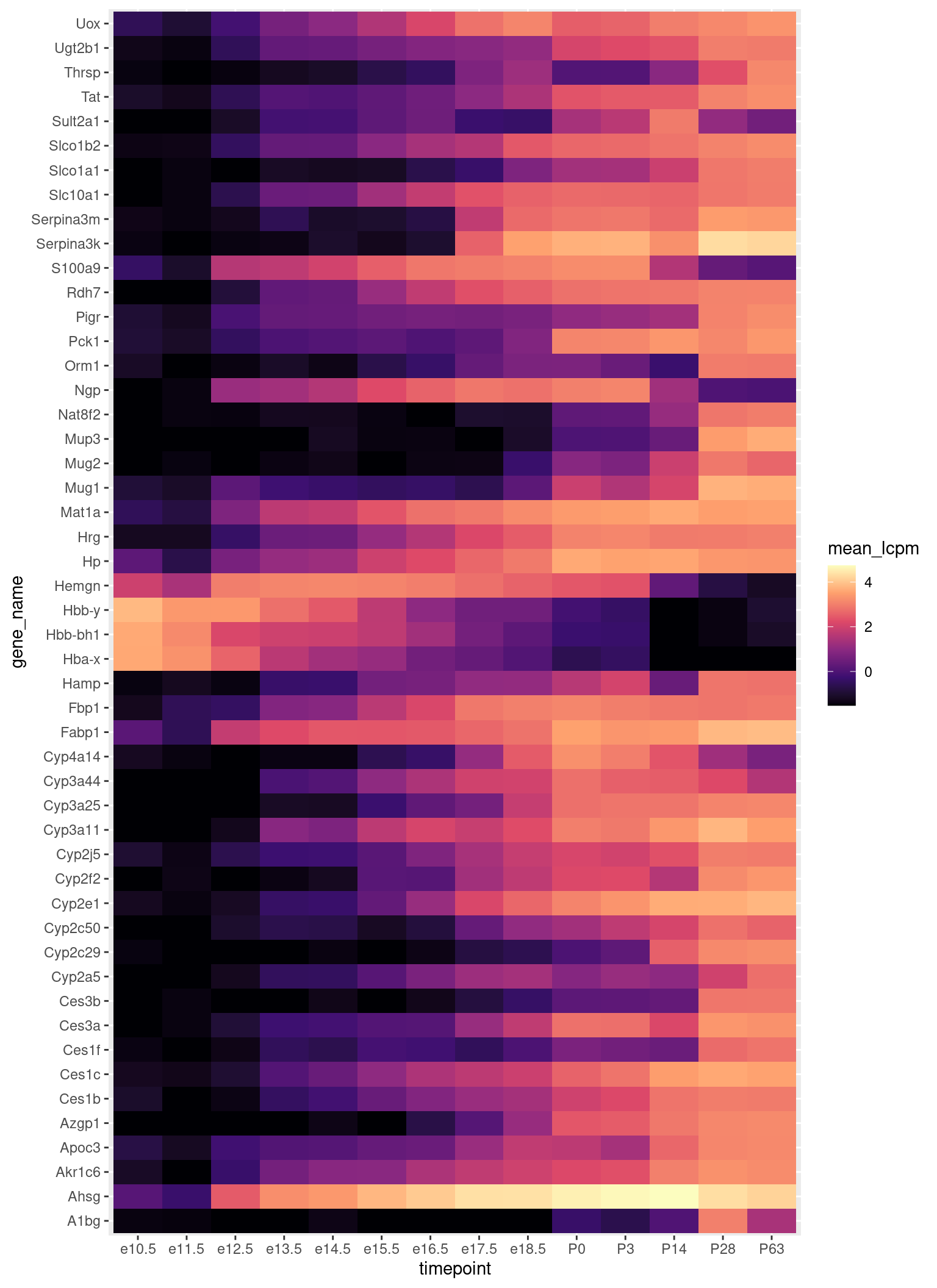
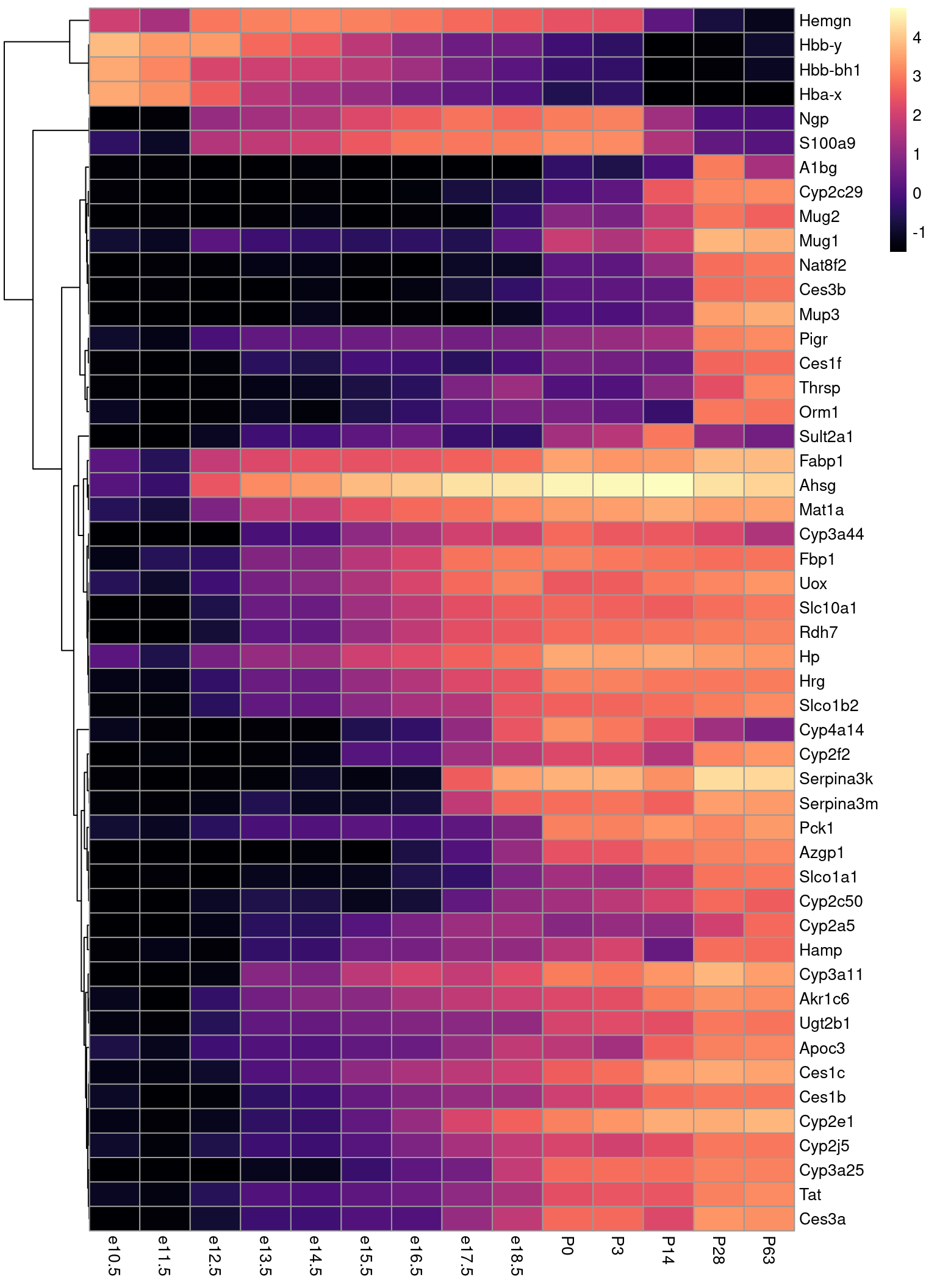
Hier ist eine andere Darstellung derselben Daten, nämlich mit geom_tile statt geom_line. Wir tauschen y-Achse gegen Farbe: die gene unterscheiden wir nun, indem wir sie untereinander anordnen, und die Expressionsstärke stellen wir durch die Farbe dar.
Etwas unübersichtlich ist es immer noch. Es hilt, die Gene anders zu sortieren.
Es gibt spezielel Funktionen um in solchen Heatmaps die Zeilen doer Spalten so zu sortieren, dass man besser sehen kann, was sich ähnelt. Wir werden das Paket pheatmap (pretty heatmaps) benutzen:
library( pheatmap )
Während wir ggplot mit geom_tile eben die daten in einer langen Liste übergeben haben, möchte pheatmap sie als Matrix. Die Zeitpunkte sollten also nicht untereinander, sondern nebeneinander stehen. Wir benutzen pivot_wider hierzu, und wandeln dann noch alles zu einer Matrix um, die die Gen-Namen als Zeilen-Namen hat:
Die Funktion pheatmap hat Dutzende Argumente, um das Aussehen der Heatmap anzupassen. Wir haben hier nur einige verwendet.
Das erste Argument ist die Matrix mit den Daten. Mit cluster_cols=FALSE und cluster_rows=TRUE stellen wir klar, dass die Reihenfolge der Spalten unverändert aus der Matrix übernommen werden sollen, während die Spalten durch sog. Clustering umgeordnet werden sollen. Zuletzt wählen wir mit color = viridis::magma(100) die Farbskala aus: Wir möchten die “Magma”-Palette aus dem “viridis”-Paket in 100 Abstufungungen verwenden. Mehr zu den Viridis-Paletten fidnen Sie hier.
Beim Clustering wird zunächst bestimmt, wie “ähnlich” die Zeilen zueinander sind: Zwei Zeilen gelten als “nah beieinander”, wenn sie einen hohen Korrelationskoeffizienten haben. (Zum Korrelationskoeffizienten später mehr.) Dass zur Beurteilung der Ähnlichkeit der Korrelationskoeffizient verwendet werden soll, wird durch clustering_distance_rows = "correlation" festgelegt.
Hier wird sog. hierarchisches (oder: agglomeratives) Clustering verwendet, um Gruppen ähnlicher Zielen nebeneinander zu setzen. Näheres heirzu findet sich hier auf Wikipedia.
Beurteilung
Mit der verbesserten Heatmap können wir jetzt einiges ablesen: Die Gene, die nur im Fötus verwendet werden, haben alle mit fötalem Hämoglobin zu tun. (Erinnern Sie sich, die Blutbildung im erwachsenen Säugetier zwar nur im Knochenmark erfolgt, im Embryo aber auch in Leber und Milz, und im Fötus ein anderes Hämoglobin als im ausgewachsens Tier verwendet wird. Siehe auch hier.)
Wir können auch Gruppen von Genen erkennen, die erst im reiferen Fötus aktiv werden, und einige, die sogar erst im ausgewachsenen Tier aktiv werden. Sie werdn sicher erkennen, dass wir viel über die Leber lernen können, wenn wir solche Plots im Detail studieren.
Reproduzierbarkeit
Erstaunlich ist das Gen Sult2a1, das scheinbar nur zu einem Zeitpunkt, nämlich 14 Tage nach Geburt (P14) hoch exprimiert ist, und dann wieder abnimmt. Stimmt das, oder ist hier vielleicht ein einzelnes der 4 Replikate ein Ausreißer und hat den Mittwelwert für diesen Zeitpunkt hoch gezogen?
Wir prüfen das, indem wir auf die ursprünglichen, noch nicht über Replikate gemittelten Daten zurück gehen und das Gen plotten:
Joining with `by = join_by(sample)`
Joining with `by = join_by(gene_id)`
Warning: Transformation introduced infinite values in continuous y-axis
Wir erkennen: Das das Gen langsam ansteigt, und zum Zeitpunkt P14 maximal ist, da sind sich alle Replikate einig, auch zu dem Abfall kurz vor Geburt. Der Effekt im höheren Alter ist aber zwischen Replikaten sehr variabel.
Wir möchten aber auch nicht zu viele Plots ansehen müssen. Daher gibt es statistsiche Techniken, um automatisiert zu beurteilen, für welche Gene Effekte als signifikant gesehen werden dürfen, da alle Repplikate ein konsitentes Bild zeigen, und wo die Daten unklarer sind. Dies wird als “differential expression calling” oder als Ermittlung von “differentially expressed genes” (DGE) bezeichnet. Bei einem einfachen Vergleich zwischen zwei Gruppen handelt es sich dabei um Weiterentwicklungen aus dem t-Test heraus. Für kompliziertere Situationen, wie die Zeitreihe hier, ist dies aber etwas komplizierter. Eventuell werden wir in einer späteren Vorlesung auf Details eingehen können.
Proben-Korrelation
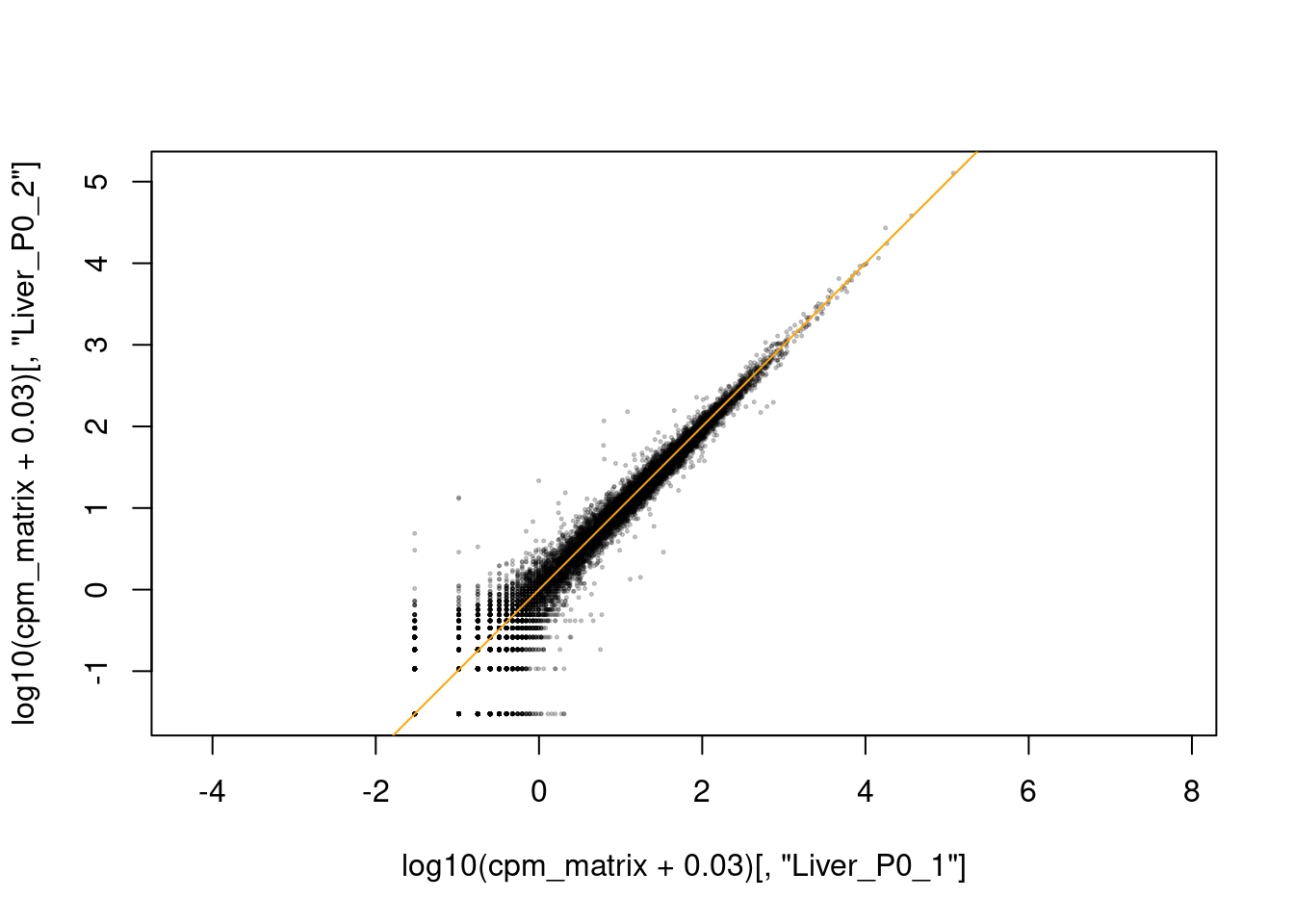
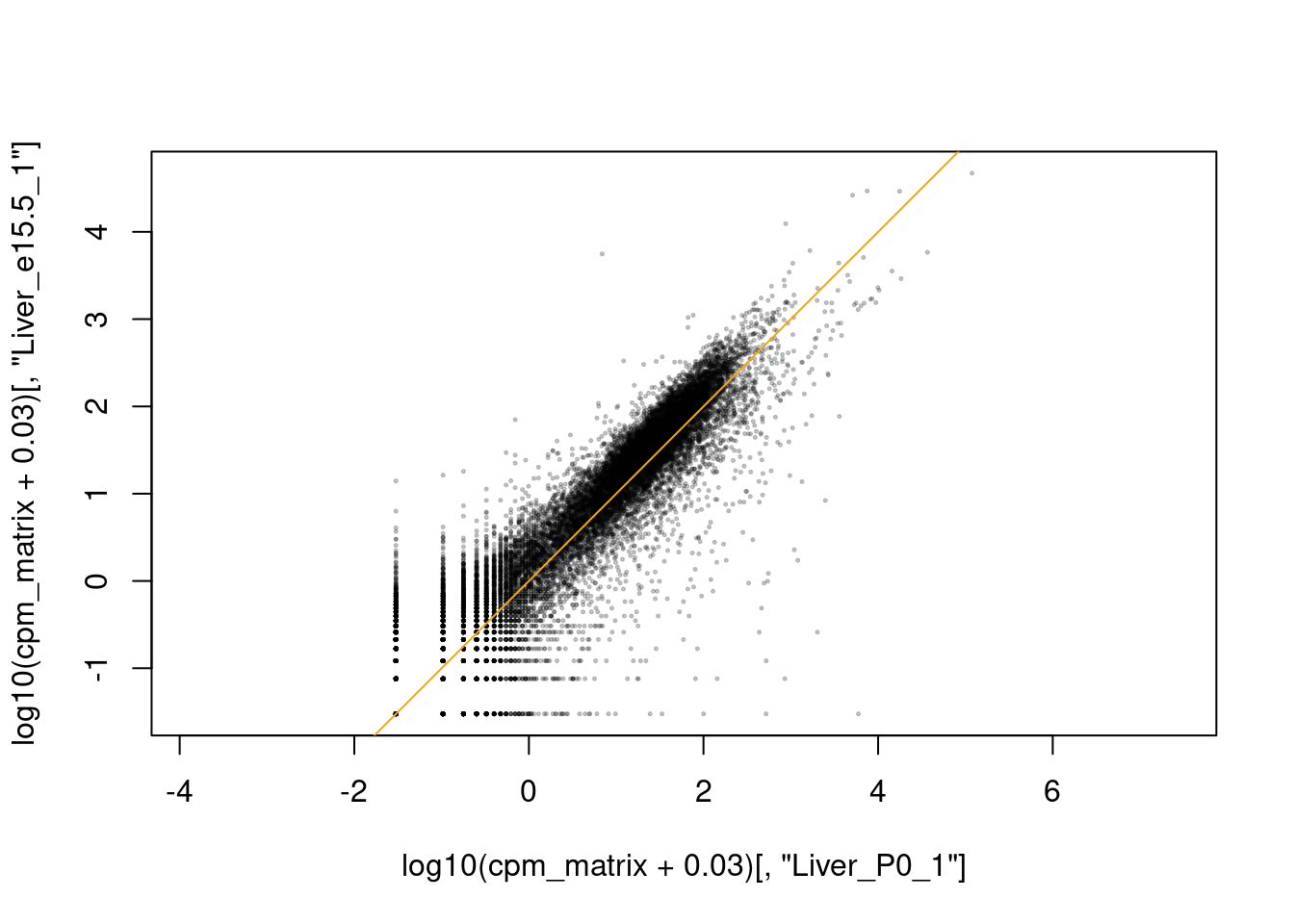
Die Frage, ob die Replikate gut miteinander übereinstimmen, können wir auch beurteilen, indem wir sie gegeneinander plotten. Wir verwenden hierzu unsere Expressionsdaten in Matrixform (siehe oben), logarithmiert:
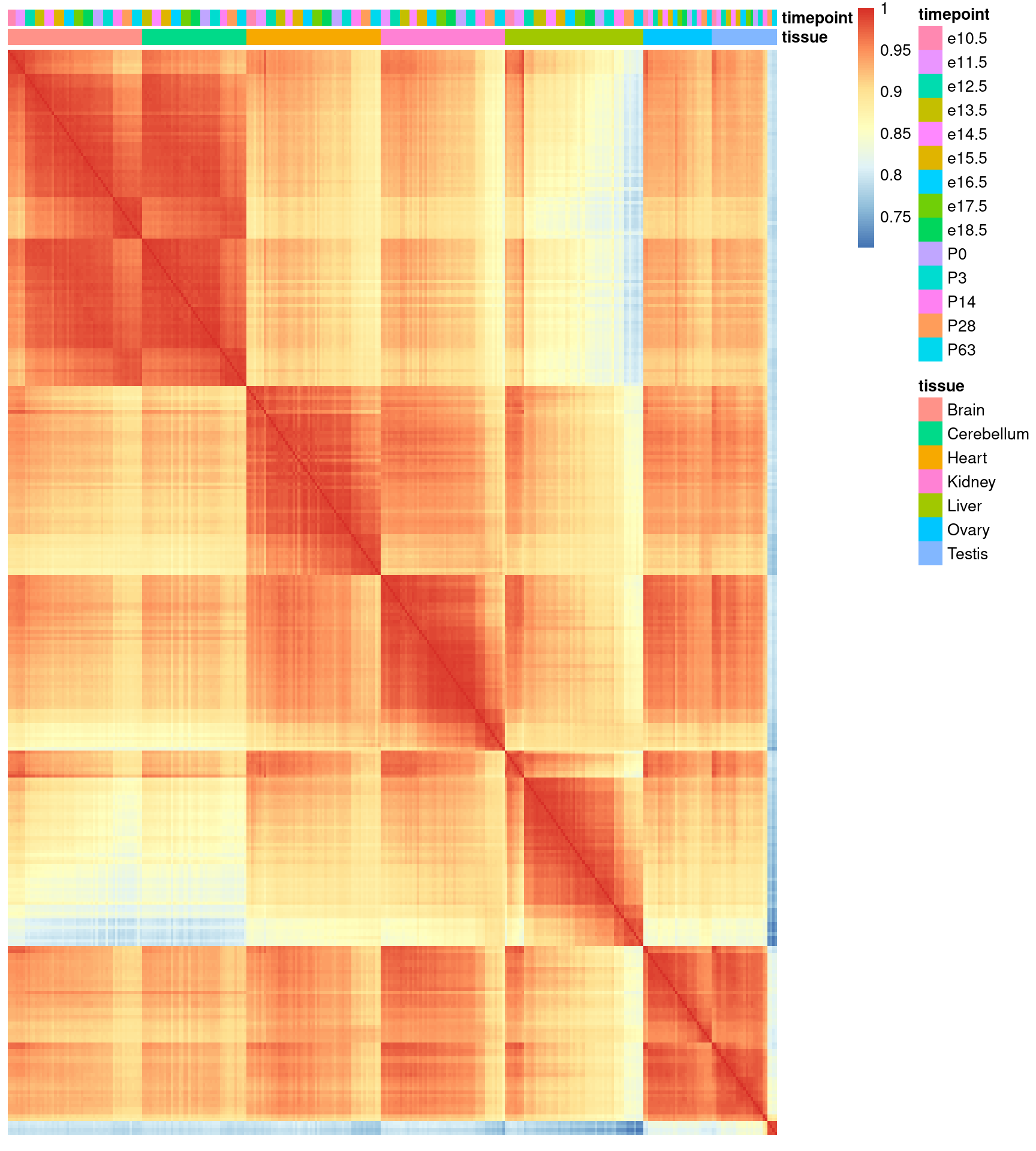
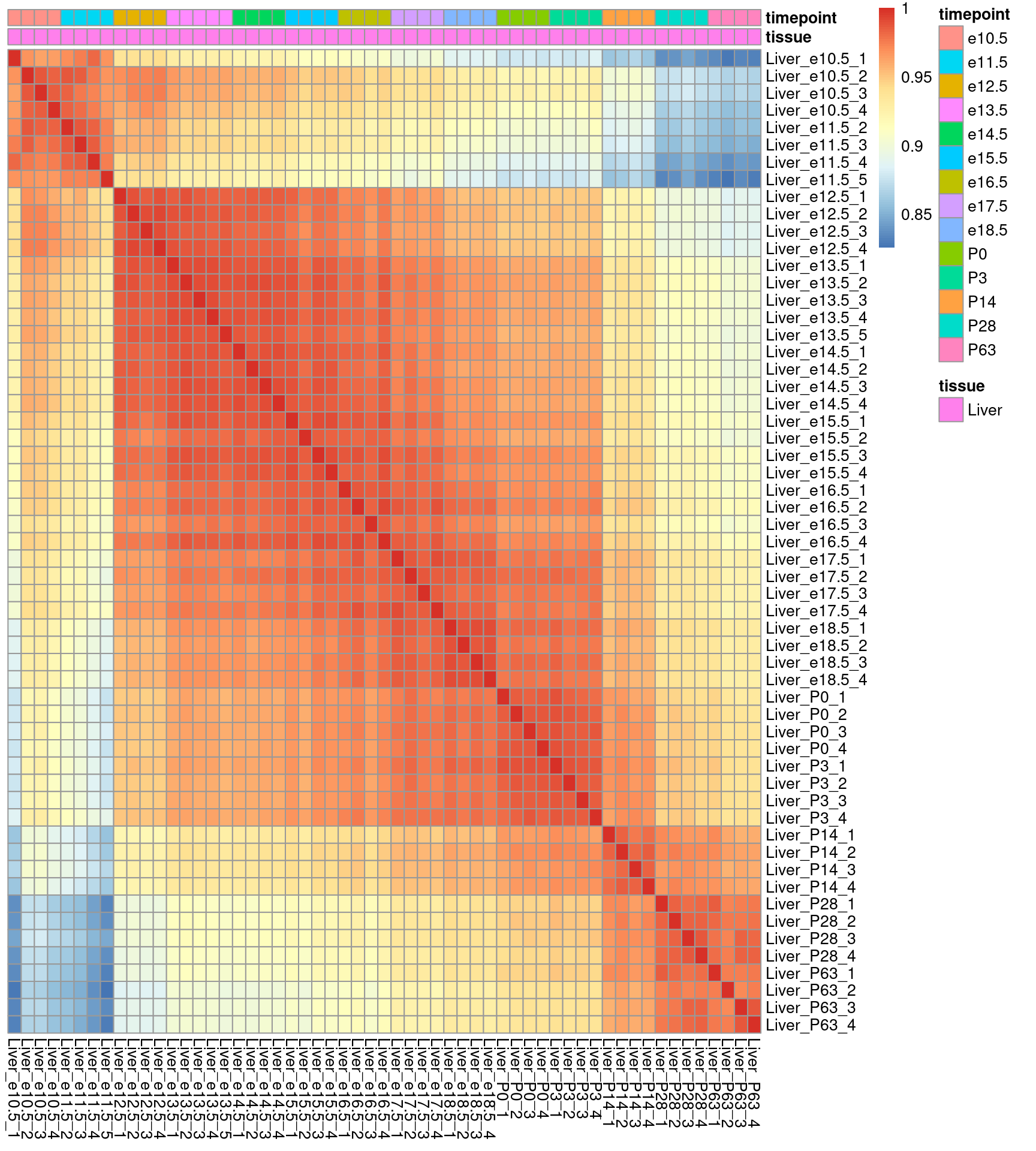
Man kann der Funktion cor auch statt zweier Spalten die ganze Matrix übergeben, und erhält dann eine sog. Korrelationsmatrix mit der Korrelation aller Spalten zu allen Spalten. Diese matrxi können wir dann mit pheatmap plotten:
Hieraus können wir nun ablesen, zu welchen Zeitpunkten sich das Leber-Transkriptom besonder stark ändert.
Aber wir erkennen auch Probleme mit der Probenqualität. Einige Proben unterscheiden sich von ihren replikaten deutlich stärker als anders, z.B. die allererste Probe.
Gewebespezifische Gene
Weiter oben haben wir gefragt, welche Gene in der Niere besodners stark exprimiert sind, konnten aber mit den gefunden Genen nicht viel anfangen. Daher stellen wir die Frage nun etwas anders:
Welche Gene sind besonders spezifisch für die Niere? Wir werden diese Frage für ausgewachsene Mäuse beantworten, indem wir uns auf die Proben von Zeitpunkt P28 beschränken.
Zunächst nehmen wir die vier P28-Proben von der Niere und bestimmen für jedes Gen den jeweils kleinsten der vier CPM-Werte:
Wenn wir diese beiden Tabellen nun nebeneinander setzen, können wir die Gene bestimmen, die in der Niere viel höher sind als in all den anderen Proben: