Vor einigen Jahren hat die Gruppe von Henrik Kässmann am ZMBH folgendes Paper veröffentlicht:
M. Cardoso-Moreia et al.: Gene expression across mammalian organ development. Nature 571:505 (2019).
Darin haben Sie Gewebeproben von 7 Organen von 7 Säugetier-Species zu jeweils mehreren Zeitpunkten der Embryogenese sowie vom heranwachsenden und ausgewachsenen Tier genommen. Zweck war es, zu sehen, wie sich die Expression aller Gene zwischen den Organen unterscheidet, wie sie sich während der Entwicklung des Tiers mit der Zeit ändert, und wie ähnlich sich diese Muster zwischen evolutionär verwandten Species ist. Insgesamt wurde RNA-Sequenzierung für 1xx Proben durchgeführt.
Wir werden zunächst anhand der Daten einer einzelnen Probe nachvollziehen, wie solche Daten zu Analyse vorbereitet werden (preprocessing) und dann einige einfache Analysen mit der Expressionsmatrix des gesamten Datensatzes durchführen.
Als Beispiel-Datensatz für die Präprozessierung verwenden wir die Sequenzierdaten von der Leber-Probe einer neugeborenen Maus.
Dem Anhang des Papers (Abschnitt “Data availability”) entnehmen wir, dass die Daten zur Maus auf dem ArrayExpress-Repository unter der Acession-Nummer E-MTAB-6798 hinterlegt ist. Dort finden wir in der “factor table” in der Zeile zu “liver” / “postnatal day 0” die Information, dass es vier Proben gibt mit den Acccessions ERS2492445 bis ERS2492448. Wir nehmen die erste Probe. Nach etwas Suchen finden wir den Link zu den Rohdaten:
Bei der Sequenzanalyse, wie auch bei vielen anderen Aufgaben ind er Bioinformatik, arbeiten wir an der Unix-Kommandozeile. Für eine ausführliche Einführung hierzu haben wir leider nicht die Zeit; es gibt aber viele Kurse hierzu, auch an der Uni Heidelberg.
Stattdessen führe ich einfach vor, wie man arbeitet, so dass Sie einen ersten Eindruck haben, auch wenn Sie die Details nicht alle werden nachvollziehen können. Ich werde auf unserem Instituts-internen Server “papagei” arbeiten. Damit Sie auch sehen, wie man die nötige Software installiert, erstelle ich innerhalb des Servers einen sog. Container, der eine anfangs noch “leere” Installation von Ubuntu Linux enthält.
docker run -it ubuntu
Zunächst installiere ich einige Werkzeuge, die ich später brauchen werde. (Normalerweise sollten diese schon da sein, aber in diesem Dockerr-Image fehlen sie.)
# Lade aktuelle Liste automatisch installierbarer Programme herunterapt update# Installier Programmeapt install wget less zip python3
Ich erstelle zunächst mit mkdir (“make directory”) ein Verzeichnis (einen Ordner, ein directiory), in dem wir unsere Daten ablegen werden und wechsele mit cd (“change directory”) in dieses:
>Mus musculus general transcription factor III C 1 (Gtf3c1), mRNA
Sequence ID: NM_207239.1 Length: 6891
>Mus musculus general transcription factor III C 1, mRNA (cDNA clone MGC:92928 IMAGE:6853285), complete cds
Sequence ID: BC067041.1 Length: 6891
Range 1: 5229 to 5329
Score:137 bits(74), Expect:3e-28,
Identities:92/101(91%), Gaps:0/101(0%), Strand: Plus/Minus
Query 1 CGCCTGTCCGCTATCTTCTCCAAGGCGAAGTACTGCCTGTTCAGCTTCTCCTTGTCAATC 60
||||||||||||||||||||||||||| || |||||||| ||||||||||||||||||||
Sbjct 5329 CGCCTGTCCGCTATCTTCTCCAAGGCGGAGAACTGCCTGCTCAGCTTCTCCTTGTCAATC 5270
Query 61 CCGAAGCAGTCAACAACTACTACAGCCTCCAGGTTCTTTAG 101
|||||||||||| || || ||||||||| |||| ||| |||
Sbjct 5269 CCGAAGCAGTCAGCAGCTGCTACAGCCTGCAGGATCTCTAG 5229
Dieser zufällig ausgewählte Read entstammt also wohl einem Fragment der mRNA des Gens Gtf3c1, das für eine Untereinheit des Transkriptionsfaktors IIIC kodiert.
Wir können zählen lassen, wie viele Zeilen die Datei enthält:
Da jeder Read 4 Zeilen hat, enthält die Datei also 19.306.170 Reads.
Short-read alignment: Aligner
BLAST ist zu langsam um so viele Reads zu alinieren. Wir verwenden Hisat2, beschrieben in diesem Ppaper:
Kim et al.: Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology, 37:907 (2019). https://doi.org/10.1038/s41587-019-0201-4
Auf der Webseite finden wir Links um Hisat2, Version 2.2.1, für Linux oder MacOS zu installieren. Wir laden die Datei mit wget herunter, entpacken sie.
cd ..wget--content-disposition https://cloud.biohpc.swmed.edu/index.php/s/oTtGWbWjaxsQ2Ho/downloadunzip hisat2-2.2.1-Linux_x86_64.zip
Genom-Index
Nun brauchen wir ein Assembly des Maus-Genoms, also eine Textdatei mit der vollständigen Sequenz aller Chromosomen der Maus. Wir finden so etwas zum Beispiel auf EnsEMBL, genauer, auf der Ensembl-FTP-Seite.
Dort finden wir u.a. diese Datei hier enthält die 22 Sequenzen, nämlich die Autosomen 1 bis 19, die Sex-Chromosomen X und Y, sowie das Genom der Mitochondrien (MT): https://ftp.ensembl.org/pub/current_fasta/mus_musculus/dna/Mus_musculus.GRCm39.dna.toplevel.fa.gz
Jede Sequenz beginnt mit einer Zeile, die mit > beginnt und dann den Namen der Sequenz (1, 2, 3, …, 19, X, Y, MT) enthält, dann ein Leerzeichen und dannn einige weitere Information. Ab der nächsten Zeile beginnt dann die eigentliche Sequenz, die sich über sehr viele Zeilen erstreckt, bis schließelich eine Zeile mit > am Anfang den Beginn des nächsten Chromosoms ankündigt.
Hisat2 kann mit einer solchen sog. FASTA-Datei nicht direkt arbeiten. Es muss erst aus dieser Datei einen sog. Index erstellen, d.h., es muss sich das Genom so aufbereiten, dass es schnell und effizient darin suchen kann. Man bunutzt daszu das Programm hisat-build und übergibt ihm den namen der FASTA-Datei mit den Genom-Sequenzen, und den namen, den die Index-Dateien erhalten sollen.
Dieser Schritt kann einige Stunden dauern. WIr überspringen ihn daher, denn von der Hisat2-Webseite können fertige Indexe für einige Genome herunter geladen werden.
Wir suchen den Link für den Index “grcm38”, laden die Datei herunter und entapcken sie:
wget--content-disposition https://cloud.biohpc.swmed.edu/index.php/s/grcm38/download tar xvf grcm38.tar.gz
Das gz-Archiv enthält mehrere Dateiem, die alle zusammen den Index bilden. Alle diese dateien werden in ein Verzeichnis grcm38 entpackt; die Dateinamen starten alle mit genome.
Alignment
Nun können wir das Aligment durchführen. Wir rufen hisat2 wie folgt auf:
Benutze das Programm hisat2, dass sich im Verzeichnis ../hisat2-2.2.1/ befindet.
Dann folgen die Argumente für das Programm (wie in R die Argumente für einen Funtionsaufruf, aber ohne Klammern und Kommas):
-x ../grcm38/genome: Der Genom-Index findet sich in den Dateien im Verzeichnis ../grcm38, deren Namen mit genome anfängt
-U 1743sTS.Mouse.Liver.0dpb.Female.fastq.gz: Die Reads finden sich in dieser Datei. Das “U” steht für “unpaired”, da es sich nicht um paired Reads handelt.
-p 10: Benutze 10 Prozessor-Kerne gleichzeitig
> 1743sTS.Mouse.Liver.0dpb.Female.sam: Sammle die Ausgabe von hisat2 auf und speichere sie in dieser Datei.
Alle diese Argumente, und viele weitere, können in der Dokumentation zu hisat2 nachgeschlagen werden.
Nach etwa 5 Minuten sind die 19 Millionen Reads aliniert. (Mit nur einem Prozessor hätte es 10mal so lange gedauert). Hisat2 fasst zusammen:
19306170 reads; of these:
19306170 (100.00%) were unpaired; of these:
981926 (5.09%) aligned 0 times
16550422 (85.73%) aligned exactly 1 time
1773822 (9.19%) aligned >1 times
94.91% overall alignment rate
Die Ausgabedatei enthält nun etwa 21 Millionen Zeilen, die z.B. so aussehen:
HISEQ:49:C2MJ0ACXX:1:1101:5228:58316: Das ist die ID des Reads, die vom Sequencer vergeben wurde. (Sie verrät uns, dass die Sequenz von einem Gerät des Typs “HiSeq” gelesen wurde; wir finden die Seriennummer des Sequenzierers oder der Flowcell, die Nummer der Lane, der Tile und die x- und y-Koordinate des Clusters im Tile.)
Dann folgt das sog. Flag-Field; die 0 zeigt ein normales Alignment an. 16 bedeutet z.B., dass der Read auf das reverse complement des Genoms gemappt wurde.
17 ist das Chromosom. Dor wurde die Sequenz des Reads an Position 57217281 gefunden.
60 ist ein Score, wie sicher der ALigner ist, dass dies der richtige Ort ist.
Der sog. CIGAR-String 52M1126N49M zeigt an, dass ab der eben genannten Position die ersten 52 Basen des Reads alinieren, dann werden 1126 Basenpaare übersprungen (vermutlich ein Intron) und dann kommen die restlichen 49 Basend es Reads (wohl im nächsten Exon).
Es folgt u.a. der Read selbst und der Basecall-Quality-String, übernommen aus den Rohdaten
und ein paar sog. Tags, die u.a. angeben, wie viele mögliche Alignments gefunden wurden (nur eines, NH:1) und wie viele Basen nicht gestimmt haben (number of mismatches, NM, hier 0).
Wir werfen im Ensembl-Genom-Browser einen Blick auf die Position 57,217,281 auf Chromosom 17 und die darauf folgenden 2000 Basenpaare: https://www.ensembl.org/Mus_musculus/Location/View?r=17%3A57217281-57219281
Wir merken gleich, dass dort kein Gen ist. Aber: Ensembl zeigt Version 39 der Maus-Genoms; wie haben aber GRCm38 verwendet. Ein Klick recht unten (“View in Archive”) erlaubt uns, auf Ensembl 102 (mit Version 38) zurück zu gehen: http://nov2020.archive.ensembl.org/Mus_musculus/Location/View?r=17%3A57217281-57219281
Wir sehen nun, das unser Read auf das Gen “C3” (complement component 3) fällt, und das er tatsächlich ein gut 1000 bp langes Intron überspringt.
Feature counting
Wir würden nun gerne für jeden einzelnen Read wissen, von welchem Gen er stammt.
Auch hierfür gibt es Werkzeuge, z.B.:
Liao, Smyth and Shi: featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30:923 (2014). https://doi.org/10.1093/bioinformatics/btt656
Wir suchen auch hier den Download-Link auf der Webseite für featureCount, laden das Programm herunter und entpacken es
cd ..wget--content-disposition https://sourceforge.net/projects/subread/files/subread-2.0.6/subread-2.0.6-Linux-x86_64.tar.gz/download
featureCount braucht eine Annotations-Datei im GTF-Format. Wir finden auch diese auf der FTP-Seite von Ensembl. Dabei achten wir darauf, dem Archiv eine Version zu entnehmen, die zu GRCm38 passt:
Dies bedeutet, das sich auf Chromosom 17, an Position 57219966 bis 57219972, ein Exon des Gens mit Ensembl-ID “ENSMUSG00000024164” und Gen-Symbol “C3” befindet.
Speziell diese exon-Zeilen verwendet featureCounts nun, um die alinierten Reads Genen zuzuordnen.
Wir starten das Programm
cd mouse_data../subread-2.0.6-Linux-x86_64/bin/featureCounts-T 10 -a ../Mus_musculus.GRCm38.102.gtf.gz -o mouse.counts 1743sTS.Mouse.Liver.0dpb.Female.sam
Hier haben wir dem Programm featureCounts folgende Argumente übergeben: - -T: Verwende 10 Prozessorkerne - -a: verwende diese GTF-Datei - -o: Schreibe das Ergebnis in diese Datei - zuletzt folgt der Name der SAM-Datei mit den alinierten Reads
Nach wenigen Sekunden berichtet featureCounts, dass es 66.4% unserer Reads einem Gen zuordnen konnte.
In der Ausgabe-Datei mouse.counts finden wir nun für jedes Gen die Ensembl-ID des Gens
Count-Matrix
Wenn wir die FASTQ-Dateien alle 1xx Proben aliniert hätten, könnten wir sie alle zusammen an featureCounts übergeben und würden so eine große Matrix erhalten, mit einer Zeile für jedes Gen, das in der GTF-Datei erwähnt wird, und einer Spalte für jede SAM-Datei.
Das müssen wir zum Glück nicht selbst machen, denn die Autoren des Papers haben ihre Matrix zur Verfügung gestellt, nämlich hier: https://www.ebi.ac.uk/biostudies/files/E-MTAB-6798/Mouse.CPM.txt.
Vergleich der Daten
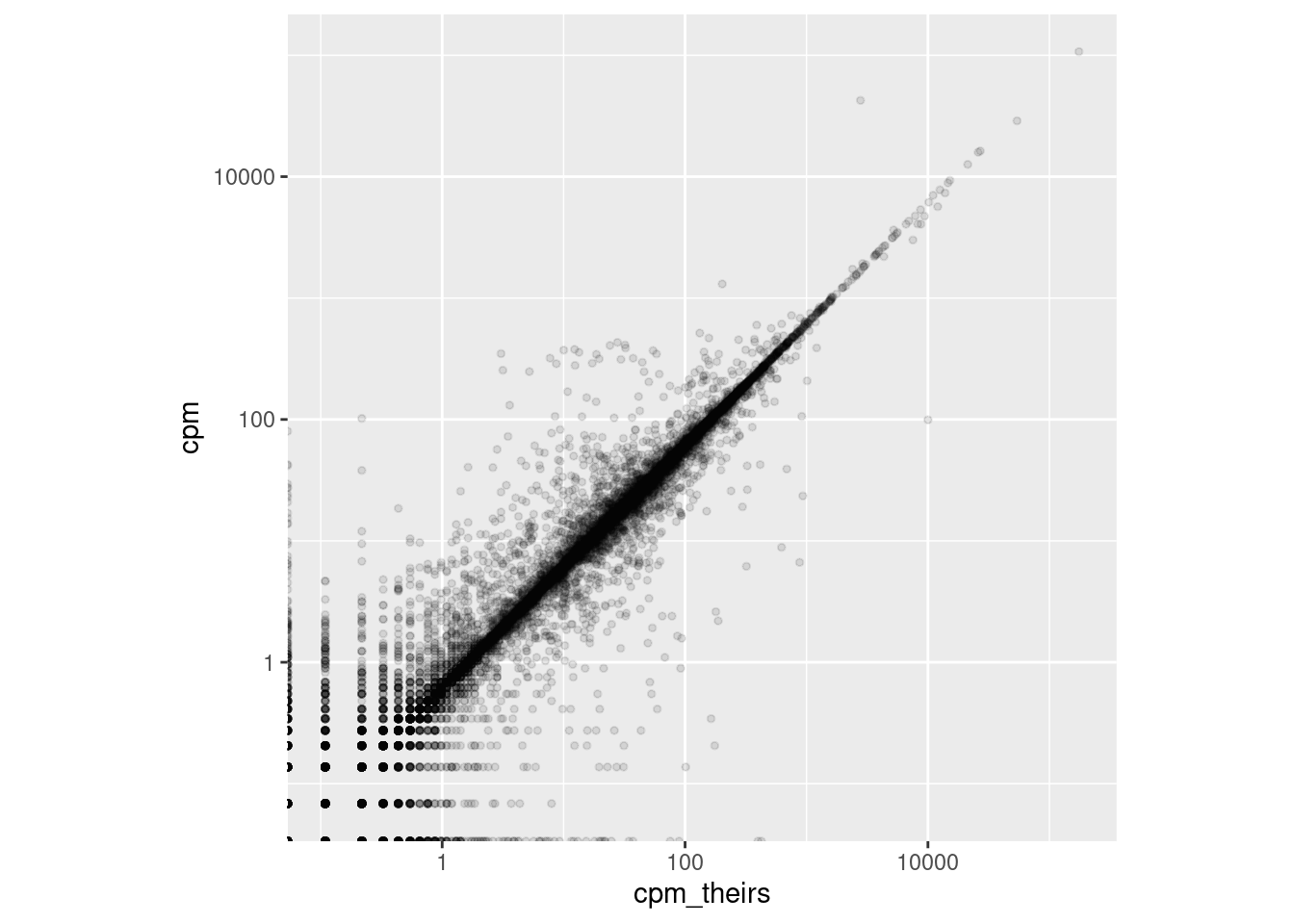
Wir verwenden nun R, um unsere Read-Zahlen mit denen der Autoren zu vergleichen.
Unsere Counts
Wir lesen zunächst unsere mit featureCounts erzeugte Datei ein
Rows: 55487 Columns: 7
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (5): Geneid, Chr, Start, End, Strand
dbl (2): Length, 1743sTS.Mouse.Liver.0dpb.Female.sam
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Wie wir sehen, haben wir einige zusätzliche Daten erhalten, nämlich zu jedem Gen die Ensembl-ID, das Chromosom, die Position von Anfang und Ende (oder mehrere, falls es Transkript-Isoforme gibt), den Strang, und zuletzt die Read-Zahl. Wir vereinfachen das:
Rows: 56305 Columns: 3
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (3): Gene stable ID, Gene name, Gene description
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Wie viele Reads man insgesamt aus einer Probe erhält, ist eine rein technische Zahl, die wenig mit der Probe zu tun hat. Es interessiert eigentlich nur, welcher Anteil dieser Reads auf jedes Gen fällt. Daher macht es Sinn, die Zahlen für die einzelnen Gene jeweils durch diese Gesamtzahl zu teilen. Da dabei aber sehr kleine Zahlen entstehen, nimmt man das Ergebnis dann üblicherweise mal eine Million und nennt den Wert “counts per million”, oder kurz “cpm”:
Für die meisten Gene sind die Werte ähnlich, aber für einige Gene gibt es deutliche Unterschiede. Vermutlich haben die Autoren einen anderen Aligner verwendet und eine andere Methode, um die Reads Genen zuzuordnen.