Anmerkung: Falls die Tabellen in den folgenden Schritten zu groß für Ihren Computer werden, können Sie die Matrix mit folgendem Code etwas verkleinern:
Der folgende Code erzeugt aus den Probennamen (Spaltennamen der Count-Matrix) eine Tabelle mit allen Proben, indem er die Spaltennamen bei den Punkten aufteilt und auf drei neue Spalten verteilt. (Da manche Zeitpunkte auf “.5” enden, und an diesen Punkten nicht getrennt werden soll, müssen wir den Trenner als kryptische sog. “Regex” angeben. sep="\\.(?!5\\.)" bedeutet: Trenne an Punkten, denen kein “5.” folgt.)
Das Format für die Zeitpunkte der Probennahme ist wie folgt zu verstehen: e13.5 bedeutet “embryonic day 13.5”, d.h. 13.5 Tage nach Befruchtung der Eizelle und P3 bedeutet: 3 Tage seit Geburt. Das fct_inorder stellt sicher, dass die Zeitpunkte später beim Plotten in derselben Reihenfolge angeordnet werden wie in den Spalten der Matrix.
Schritt 1: Pivotieren
Wandeln Sie die Count-Matrix in eine lange Tabelle um, mit einer Zeile pro Wert und Spalten “sample”, “gene_id” und “count” für die Proben-Bezeichnung (Spaltenname in der Count-Matrix), Gen-ID und Read-Count.
Schritt 2: Normalisieren
Berechnen Sie für jede Probe die Gesamtzahl der Reads, indem Sie nach Probe (“sample”) gruppieren und dann summieren. Teilen Sie die Readzahl für jedes Gen durch die Gesamzahl und multiplizieren Sie mit 1 Million. Nennen Sie die Spalte mit diesen Werten “cpm”, für “counts per million”.
Tipp: Sie brauchen kein summerize. Verwenden Sie group_by und danach ein mutate mit count / sum(count).
Schritt 3: Table-Joins
Ergänzen Sie dann die Spalten für Organ, Zeitpunkt und Replikat, indem Sie die Probentabelle per Table-Join anfügen.
Hier finden Sie eine Tabelle, die die Gen-IDs den Gen-Symbolen (Gen-Namen) zuordnet. Fügen Sie auch diese Tabelle hinzu.
Schritt 4: Plotten
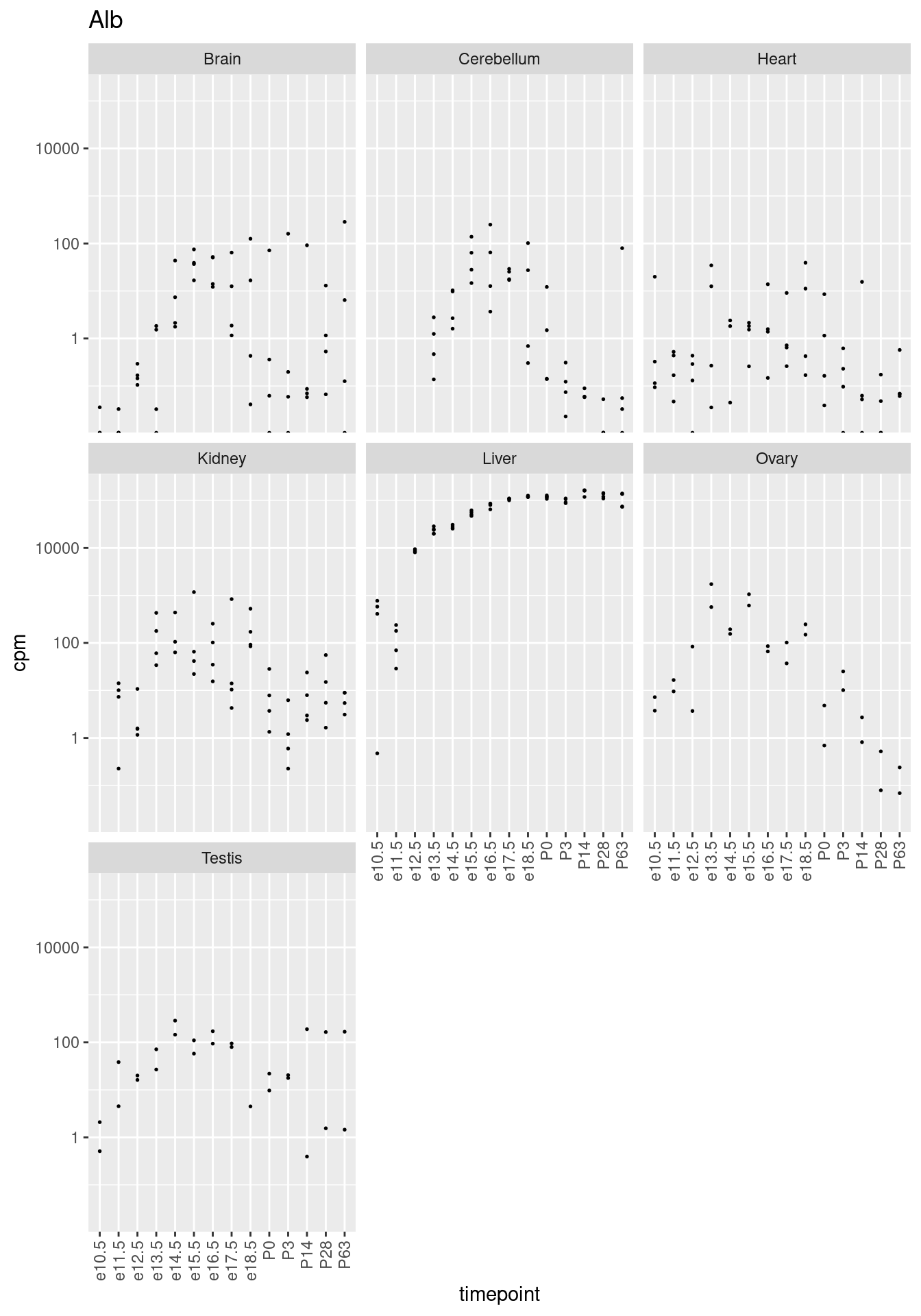
Plotten für ein Gen die Expression (CPM-Werte, mit logarithmischer Achse) gegen die Zeitpunkte, facettiert nach Organ. Unten sehen Sie zum Beispiel der Plot für das Gen “Alb”, das für das Protein Serum-Albumin kodiert.
Versuchen Sie folgende Gene:
Alb
Hbb-b1 und Hbb-bh3
Mbp
Islr2
Gpt
Schlagen Sie jeweils nach, was über die Gene bekannt ist.