RNA-Seq wird häufig angewendet, um zu untersuchen, welche Gene in ihrer Expressionsstärke von einem gegebenem äußerem Umstand abhängen. Im einfachsten fall vergleicht man dann zwei Gruppen von Proben, zum Beispiel:
Zellkulturen, die mit einem Wirkstoff behandelt wurde vs solche, die mit einer “vehicle control” behandelt wurden
Gewebeproben von erkrankten vs gesunden Patienten
Gewebeproben von Pflanzen, die bei viel Licht versus wenig Licht gewachsen sind
…
Oft ist das experimentelle Design komplizierter als ein einfacher Vergleich zweier Gruppen.
Wie bereits erwähnt, gibt es spezialisierte Software, die solche Analsyen durchführen kann. EIn Paket (an dessen Enwticklung ich beteilgt war) ist DESeq2. Es kann mit recht komplexen Designs arbeiten; hier möchte ich aber nur die Anwendung für einen einfachen Zwei-Gruppen-Vergleich vorführen.
Beispiel einer DESeq2-Analyse
Als Bespiel verwenden wir die Mausdaten von der Kaessmann-Gruppe und vergleichen die Genexpression im Herz neugeborener Mäuse mit denen in von 2 Monate alten Mäusen.
Rows: 36141 Columns: 317
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): gene_id
dbl (316): Brain_e10.5_1, Brain_e10.5_2, Brain_e10.5_3, Brain_e11.5_1, Brain...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Wir verwenden nur die Proben zu “Heart” von Zeitpunkt “P0” und “P63”, und wählen daher nur die benötigten Spalten. Dann wandeln wir die Tabelle in eine Matrix um, weil DESeq2 eine solche Matrix aus Read-Zahlen als Eingabe benötigt.:
Außerdem benötigt DESeq2 eine Proben-Tabelle, d.h. eine Tabelle mit einer Zeile pro Probe und Spalten für die Kovariaten (d.h., die Informationen, die die Unterschiede der Proben beschreiben). Unsere Tabelle ist einfach:
Einschub: DESeq2 ist als Teil des Bioconductor-Projekts veröffentlicht, welches eine Sammlung von R-Paketen füë die Biologie darstellt, die alle gewisse Mindest-Qualitäts-Standards aufweisen, und (zumindest im Prinzip) so aufeinander abgestimnmt sind, dass man sie gut kombinieren kann. Bioconductor-Pakete installiert man nicht, wie andere R-Pakete, mit install.packages, sondern mit der Funktion install aus dem Paket BiocManager. Letzteres muss man also zuerst installieren, und zwar wie gewohnt mit install.packages.
DESeq2 verwendet ein objekt-orientiertes Interface. Das bedeutet, dass alle zu einem Analyse-Projekt gehörenden Daten, zusammen mit Zwischenergebnissen, in einem einzelnen komplexen Objekt (das als eine Variable vorliegt) gebündelt sind.
Wir erstellen so in Objekt (genannt ein “DESeq Data Set”) wie folgt:
Das Objekt dds bündelt jetzt die drei Informationen, die wir übergeben haben: die Matrix mit den Read-Zahlen, die Proben-Tabelle und die sog. Design-Formel. Letztere beschreibt, wie die Spalten in der Probentabelle bei der Auswertung zu nutzen sind. Bei einem Zwei-Gruppen-Vergleich ist die Design-Formel sehr einfach, nämlich eine Tilde, gefolgt vom Namen der Spalte, die Zuordnung zu den zwei Gruppen angibt.
Die eigentlich Analyse wird dann automatisch durch die Funktion DESeq durchgeführt. Sie liefert eine Kopie des DESeq-Datasets, in das die Ergebmnisse eingefügt wurden. Wir schreiben das in die Variable zurück:
dds %>% DESeq -> dds
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
Das Ergebnis können wir nun mit der results-Funktion als Tabelle erhalten:
results(dds)
log2 fold change (MLE): timepoint P63 vs P0
Wald test p-value: timepoint P63 vs P0
DataFrame with 36141 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSMUSG00000000001 1513.35599 -0.606287 0.142385 -4.25808 2.06190e-05
ENSMUSG00000000003 0.00000 NA NA NA NA
ENSMUSG00000000028 216.41887 -2.059059 0.152941 -13.46313 2.57748e-41
ENSMUSG00000000037 101.21551 -1.800258 0.373524 -4.81965 1.43808e-06
ENSMUSG00000000049 4.79734 1.570935 1.015467 1.54701 1.21861e-01
... ... ... ... ... ...
ENSMUSG00000102128 0 NA NA NA NA
ENSMUSG00000102129 0 NA NA NA NA
ENSMUSG00000102130 0 NA NA NA NA
ENSMUSG00000102131 0 NA NA NA NA
ENSMUSG00000102132 0 NA NA NA NA
padj
<numeric>
ENSMUSG00000000001 7.19981e-05
ENSMUSG00000000003 NA
ENSMUSG00000000028 1.07213e-39
ENSMUSG00000000037 5.85795e-06
ENSMUSG00000000049 1.96935e-01
... ...
ENSMUSG00000102128 NA
ENSMUSG00000102129 NA
ENSMUSG00000102130 NA
ENSMUSG00000102131 NA
ENSMUSG00000102132 NA
results leifert das Ergebnis als eine Tabelle vom Typ DataFrame (eine Variante von data.frame, die spezifisch für Bioconductor ist). Wir wandeln diese Bioconductor-Tabelle in eine Tidyverse-Tabelle um (über den Umweg eine Base-R-Tabelle), damit wir wie gewohnt mit ihr arbeiten können:
gene_id ist die Ensembl-ID des Gens, entnommen den Rownames der Count-Matrix
baseMean ist die durschnitttliche Expression des Gens über alle Proben. Die Einheit ist diesmal nicht “counts per million” sondern “normalized counts”. Das bedeutet das für eine durschnittlich tief sequenzierte Probe eine Einheit in etwa einem Read entspricht. Genaueres siehe unten.
log2FoldChange: Das logarithmische Expressionsverhältnis zwischen den (geometrischen) Mittelwerten der normalisierten Counts, also der Logarithmus zur Basis 2 des Mittelwerts von P63 geteilt duirch den Mittelwert von P0.
lfcSE: log2 fold change standard error, d.h. der Standardfehler des log2FoldChange.
stat: die Test-Statistik (ein Zwischenwert bei der Berechnung von lfcSE und p-Wert)
pvalue: der p-Wert, mit dem die Nullhypothese “Die Mittelwerte [eigentlich: Erwartungswerte] sind gleich” abgelehnt wird.
padj: Der nachd er Methode von Benjamini und Hochberg (BH) adjustierte p-Wert. Siehe unten.
Rows: 56305 Columns: 3
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (3): Gene stable ID, Gene name, Gene description
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
res %>%left_join( gene_names ) -> res
Joining with `by = join_by(gene_id)`
Ein Blick auf ein einzelnes Gen
Wir fragen zunächst, für welche Gene wir einen besonders kleinen p-Wert erhalten haben:
Wir sehen wie die Normalisierung die Zahlen tiefer sequenzierter Proben etwas verringert bzw. weniger tief sequenzierter Proben etwas erhöht hat. In unserer früheren Analyse haben wir zu diesem Zweck durch die Gesamtzahl der Reads pro Probe geteilt und mit 1 Million multipliziert (“cpm”). DESeq2 macht etwas leicht anderes, wie wir späteer sehen werden, aber zum selben Zweck.
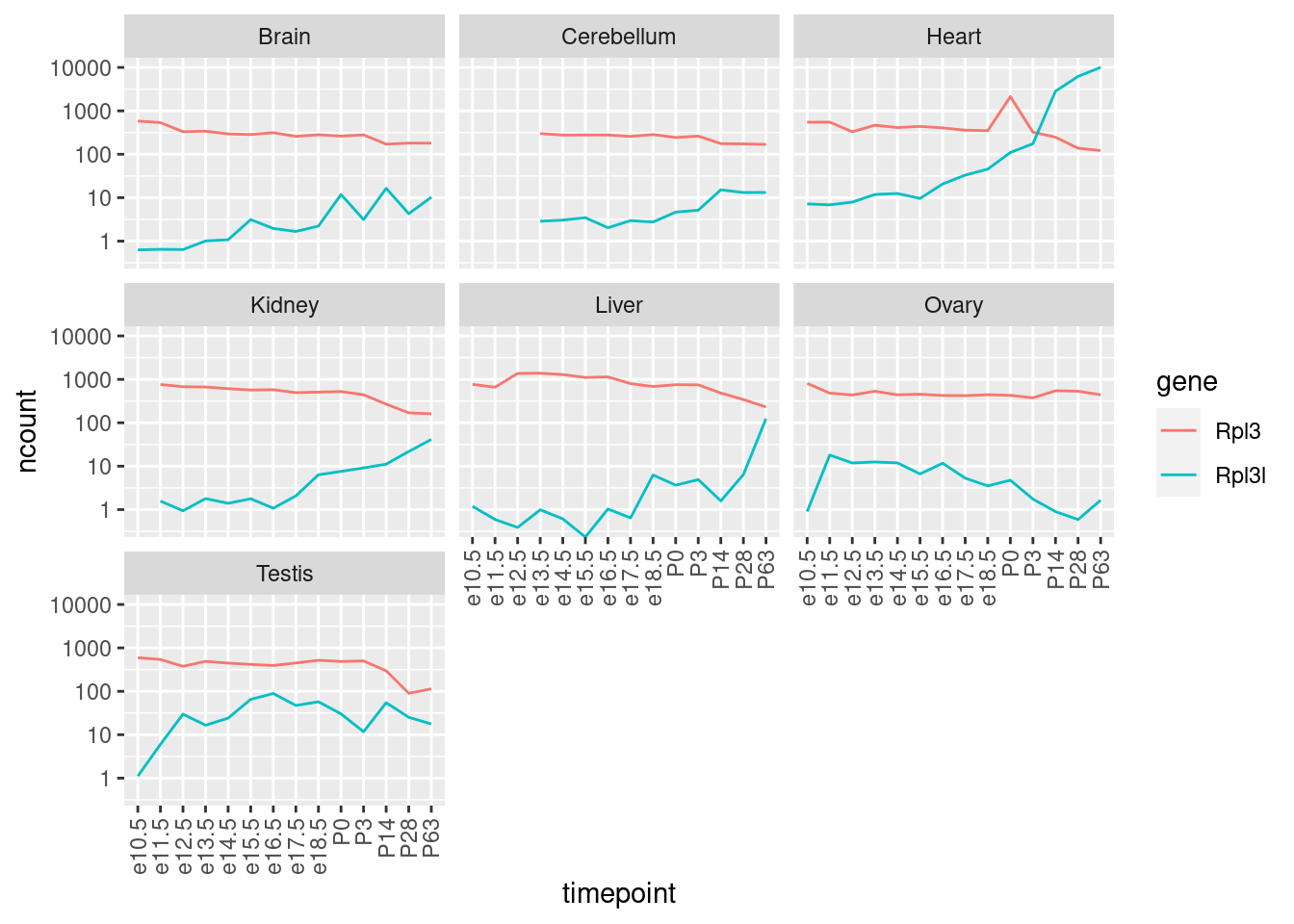
Was ist das für ein Gen? Eine kurz Google-Suche führt z.B. zu diesem Paper.
Aufgabe: Lesen Sie den Abstract des Papers und erstellen Sie dann einen geeigneten Plot, der darstellt, wie sich das Expressionsverhältnis der Gene Rpl3l zu Rpl3 über alle Zeitpunkte ändert. Wie sieht das in den anderen Geweben aus?
converting counts to integer mode
`summarise()` has grouped output by 'tissue', 'timepoint'. You can override
using the `.groups` argument.
Warning: Transformation introduced infinite values in continuous y-axis
Überblick über alle Gene
Zurück zur Ergebnis-Tabelle: Für wie viele Gene konnten wir die Nullhypothese ablehnen?
Nehmen Sie nun für einen Moment an, es gäbe in Wirklichkeit keinen Unterschied zwischen P0 und P63. Irgendjemand hat die Proben falsch beschriftet, und in Wirklichkeit sind alle 8 Proben von Mäusen desselben Alters. Die Nullhypothese, dass es keinen Unterschied zwischen den Gruppen gibt, ist also wahr. Der wahre Wert für alle log fold changes ist als 0.
Für wie viele Gene erwarten wir dann, dass das 95%-KI des LFC die Null überdeckt? – Antwort: Für 95% aller Gene.
Für wie viele Gene erwarten wir einen p-Wert unter 5%? – Antwort: Für 5% der Gene.
Für wie viele Gene erwarten wir einen p-Wert unter \(p\)? – Antwort: Für einen Bruchteil \(p\) der Gene.
Wir haben hier 23483 Gene, für die ein p-Wert berechnet werden konnte. (Die anderen sind Gene, die in allen Proben 0 Reads hatten.):
res %>%filter( !is.na(pvalue) ) %>%nrow()
[1] 23483
Für wie viele Gene würden wir einen p-Wert unter 5% erwarten, wenn die Nullhypothese für alle Gene war wäre?
23483*0.05
[1] 1174.15
Für wie viele Gene haben wir tatsächlich einen p-Wert unter 5%?
res %>%filter( pvalue <0.05 ) %>%nrow()
[1] 10643
Wenn wir diese 10643 Gene als “differentiell exprimiert” betrachten, sollten wir uns also im Klaren sein, dass darunter um die 1174 falsch positive sind, denn so viele Gene hätten wir sogar zu erwarten, wenn es in Wahrheit keinen Unterschied gäbe. Unser Anteil an Falsch-Positiven unter unseren Positiven (“Hits”, “Discoveries”) ist also etwa 11%.
1174/10643
[1] 0.1103072
Benjamini und Hochberg haben vorgeschlagen, dies so auszudrücken: Bei einer p-Wert-Grenze von 5% liegt die “false discovery rate” (FDR) bei 11%.
B&H schlagen nun weiterhin vor, zu jedem einzelnen p-Wert zu berechnen, was die FDR wäre, wenn man die Grenze genau bei diesem Wert setzen würde. Diesen Wert nennt man den BH-adjustierten p-Wert. Man kann diese Werte in R mit p.adjust( pvalues, method="BH" ) berechnen.
DESeq2 hat das schon für uns gemacht und die Ergebnisse in der Spalte padj eingetragen. Allerdings hat es vorab einige Gene entfernt, um etwas bessere adjustierte p-Werte zu erhalten (sog. independent filtering). Deshalb sind einige padj-Werte NA.
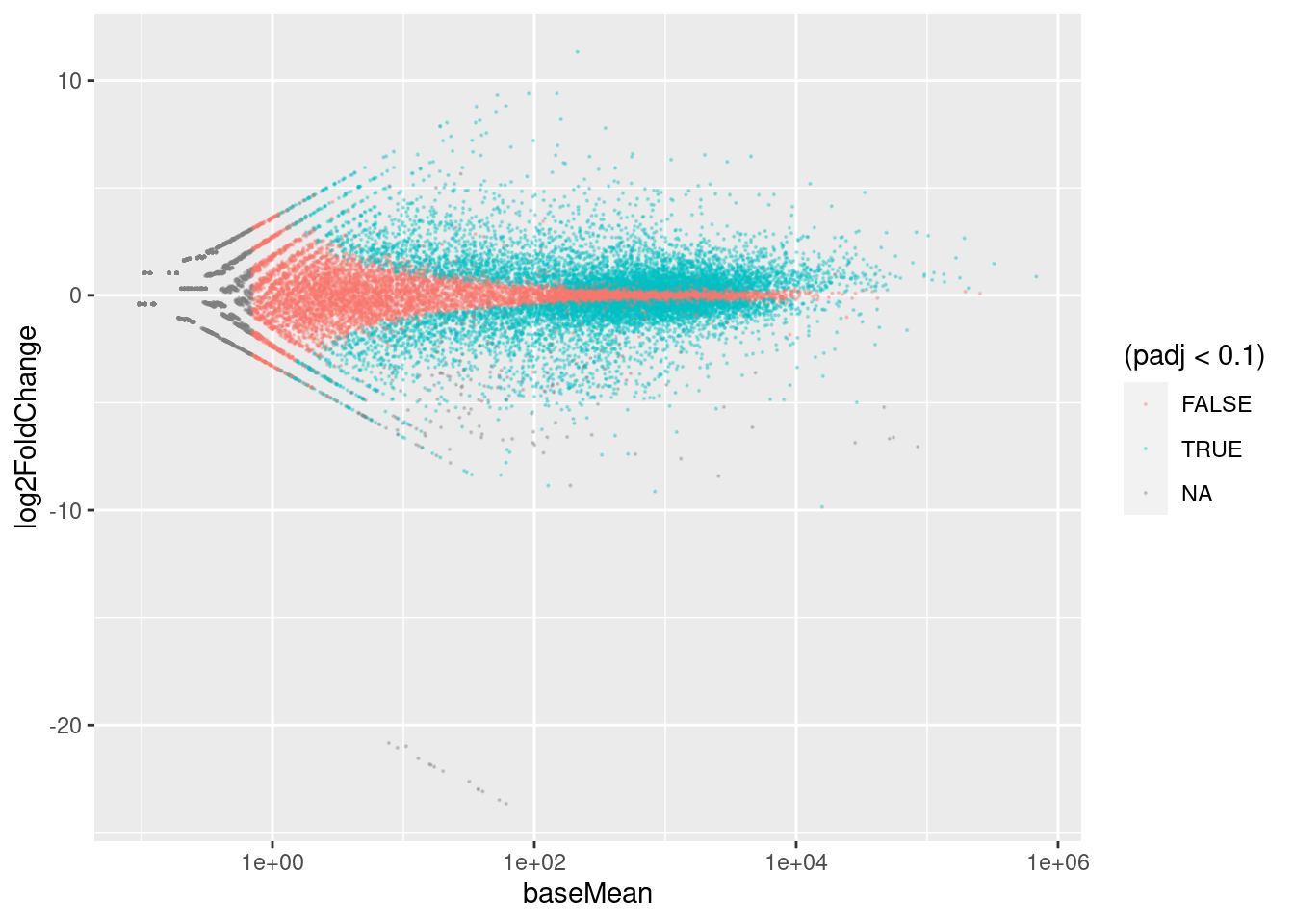
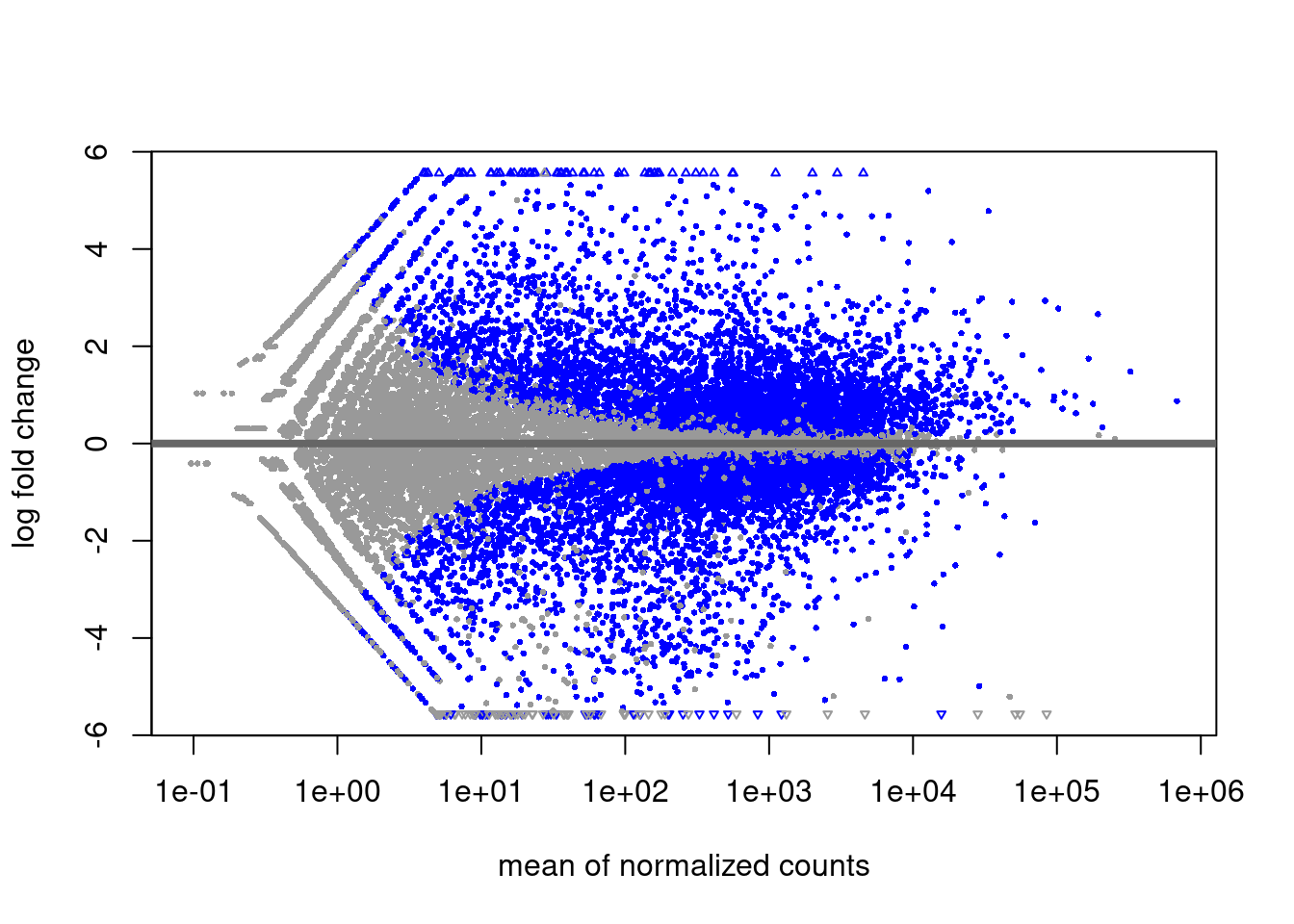
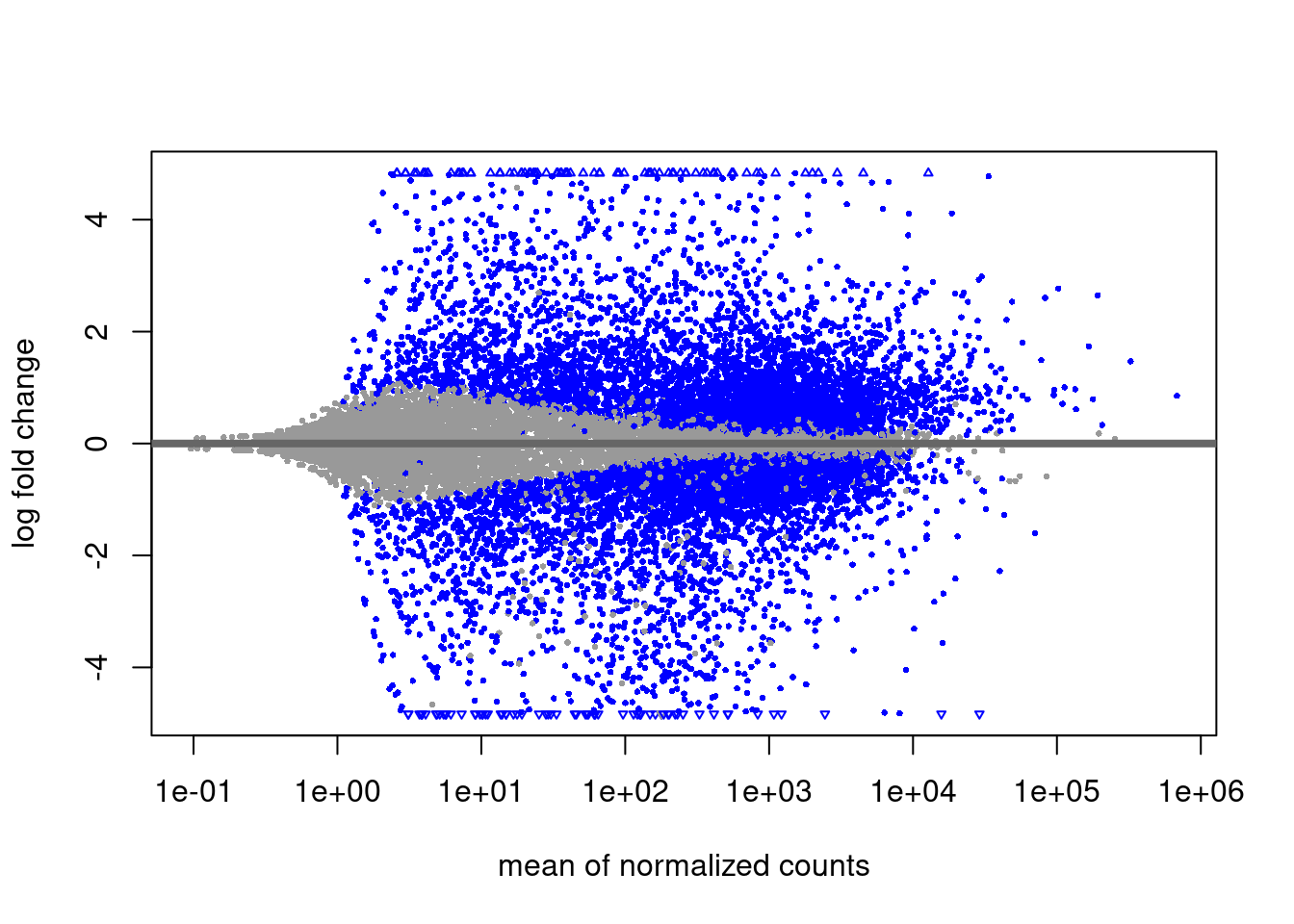
Häufig betrachtet man eine FDR von 10% als akzeptabel. Wenn wir alle die Gene als “Hits” betrachten, deren padj-Wert unter 0.1 liegt, haben wir eine p-Wert-Schwelle gewählt, die zu 10% FDR führt. Das ist, was wir oben im MA-Plot verwendet haben.
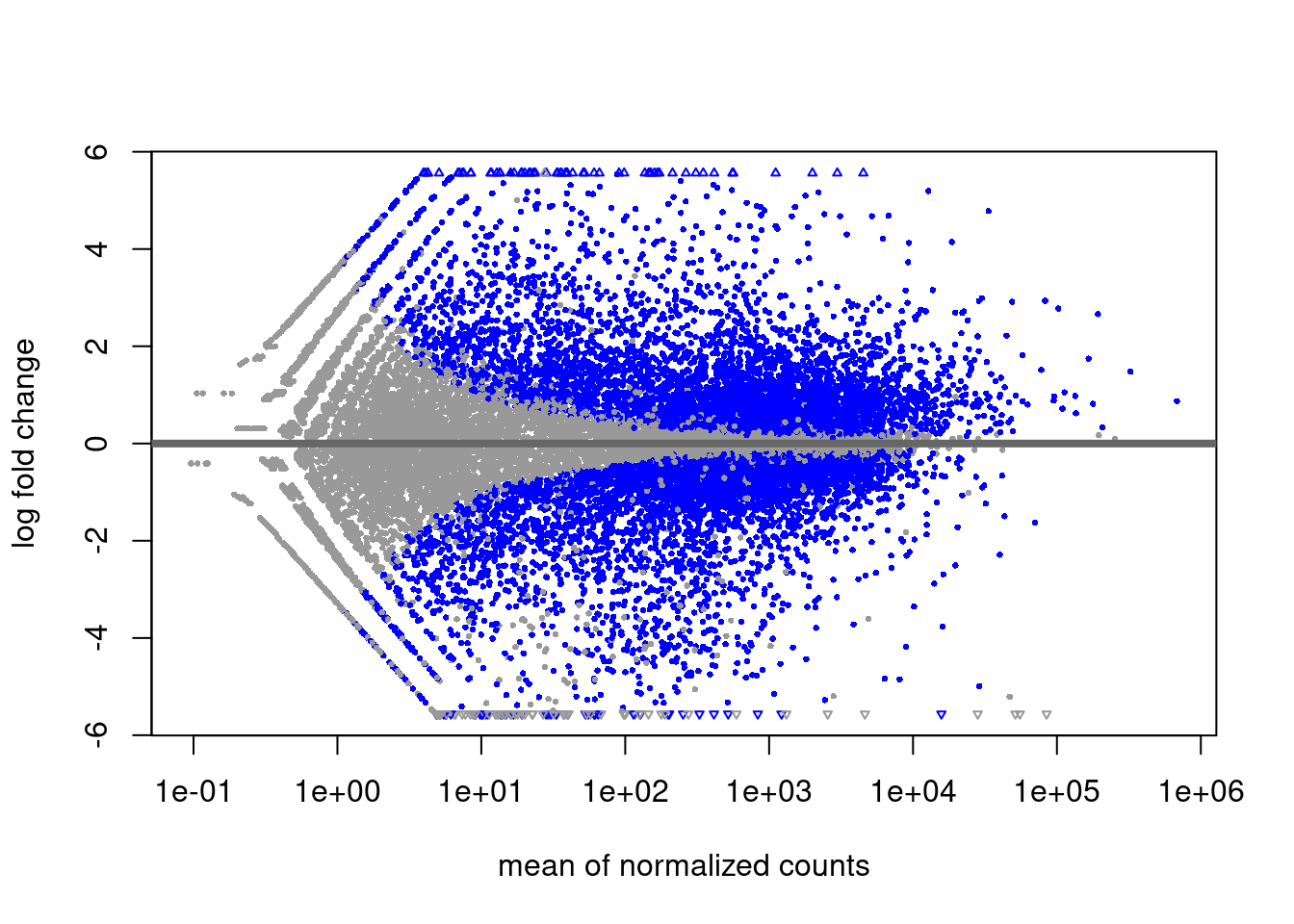
DESeq kann denselben Plot auch selbst erstellen
plotMA( dds )
Macht die Nullhypothese Sinn?
Die Genregulation in einer Zell ist hochgradig vernetzt. Kein Gen ist unabhängig von einem anderen, und wenn sich auch nur ein Gen ändert, ändern sich alle, wenn auch vielleicht nur ein ganz klein wenig.
Wir können also die Frage “Welche Gene sind unterschiedlich zwischen P0 und P63?” ohne Experiment beantworten mit: “Vermutlich alle!”
Was wir eigentlich wissen wollen ist: Welche Gene werden stärker und welche schwächer? Nur wenn das Konfidenzintervall die Null nicht überlappt, können wir sagen, ob die Änderung positiv oder negativ ist. Die wahre Bedeutung der blauen Punkte im MA-Plot ist also nicht: “Bei diesen Gene ändert sich die Expression mit dem Alter und bei den anderen nicht”, sondern: “Bei diesen Genen is der Effekt groß genug, dass wir mit einer gewissen Zuversicht sagen können, in welche Richtung sich das Gen geändert hat.”
Die FDR ist also eigentlich eine “false sign rate”: 10% der blauen Punkte sind also auf der falschen Seite der Nulllinie. Oder, anders ausgedrückt: Wenn wir das Experiment wiederholen würden, dann werden etwa 90% der blauen Gene wieder auf der selben Seite (über oder unter der x-Achse) zu liegen kommen.
Es wird aber keine Aussage über die Stärke des Effekt (Abstand zur x-Achse gemacht).
Verzerrung der Effektstärke durch Poisson-Statistik
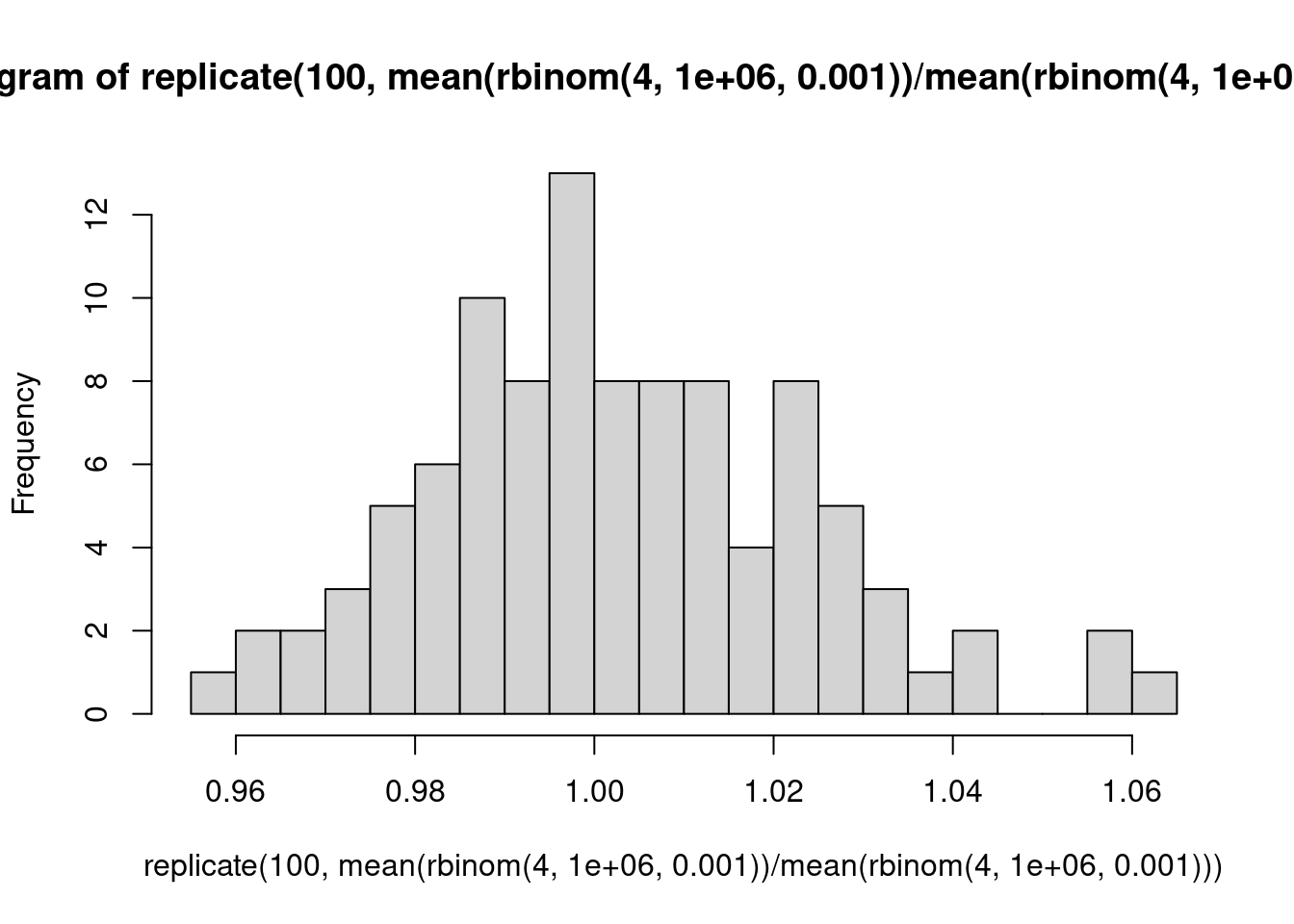
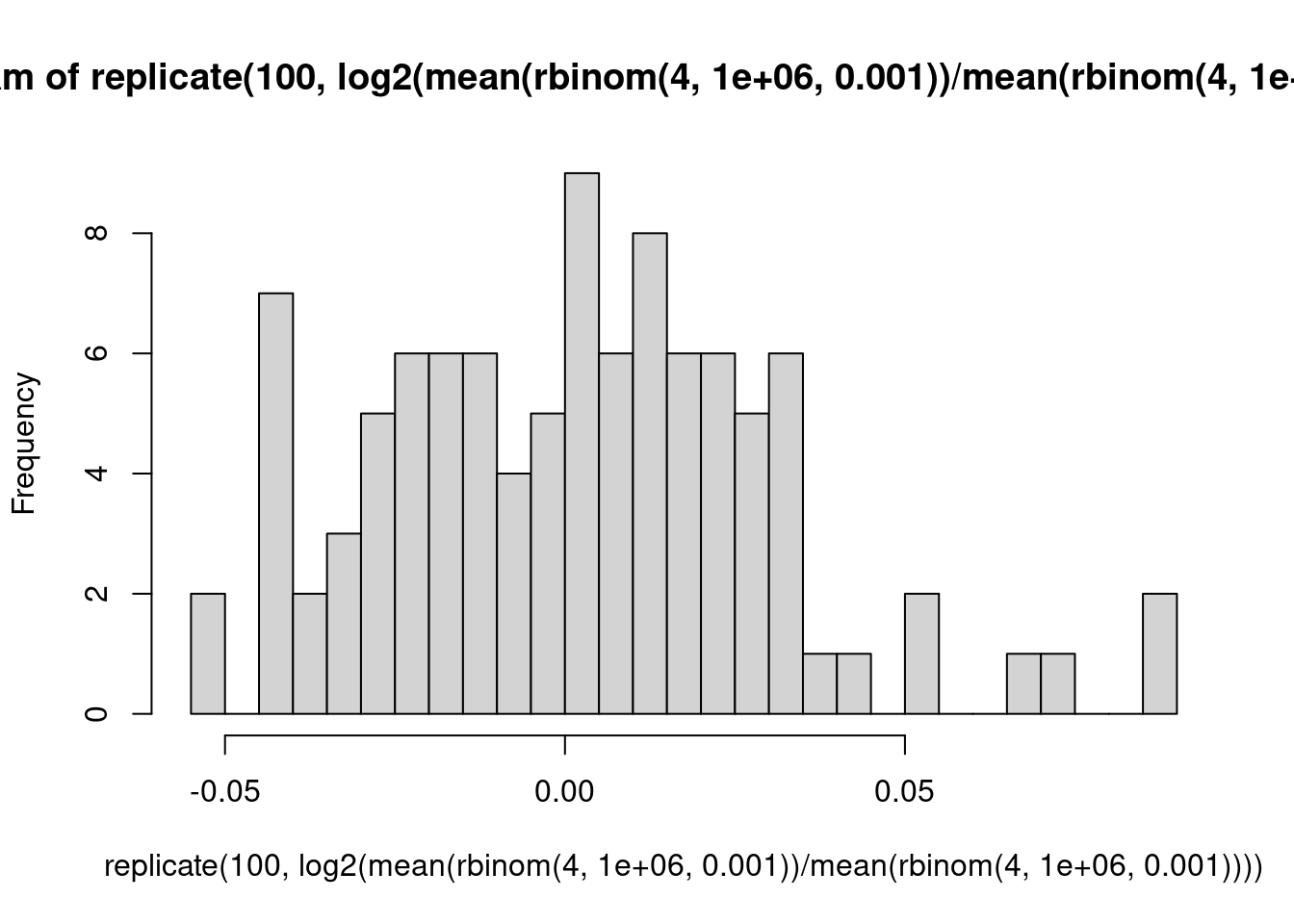
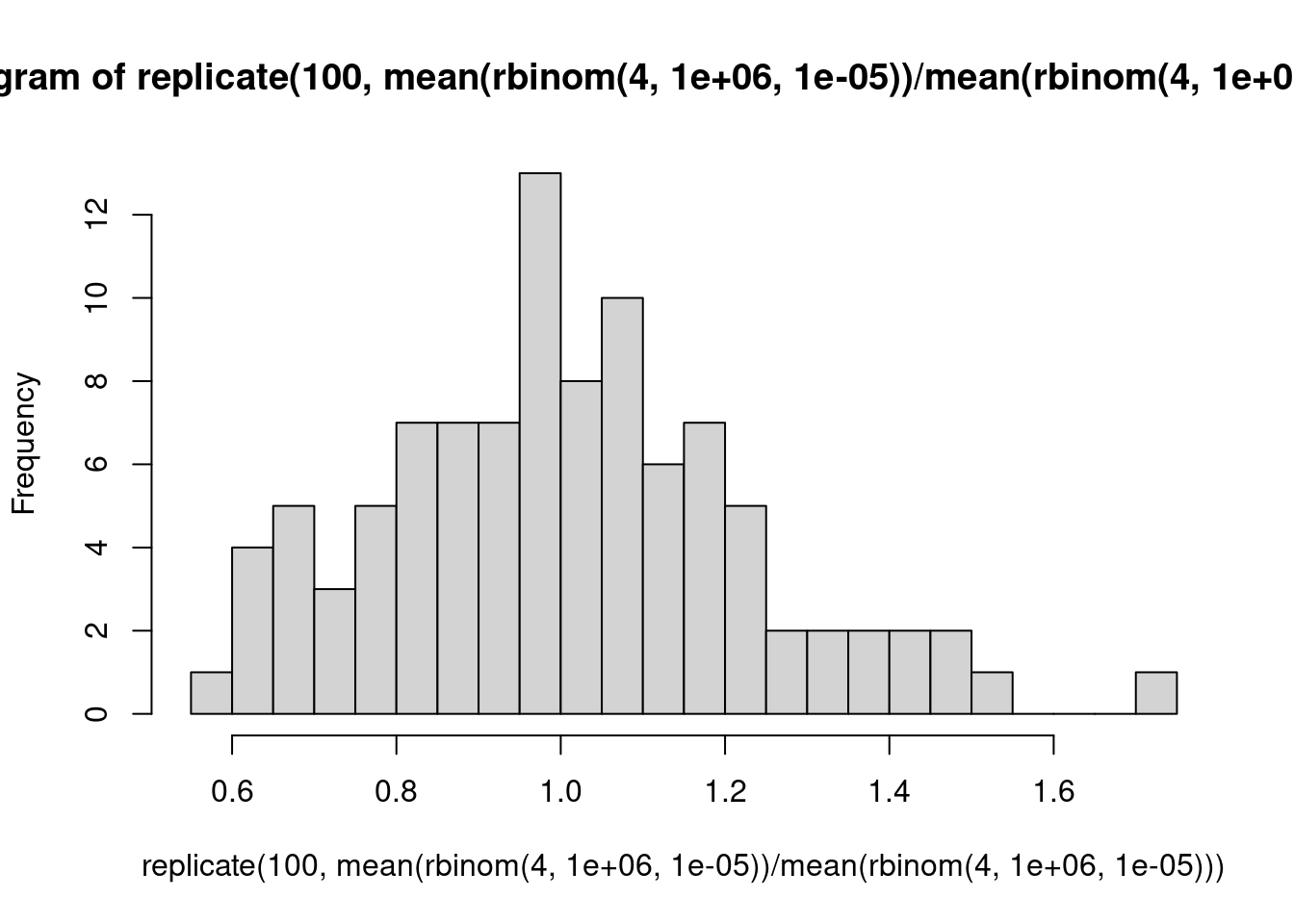
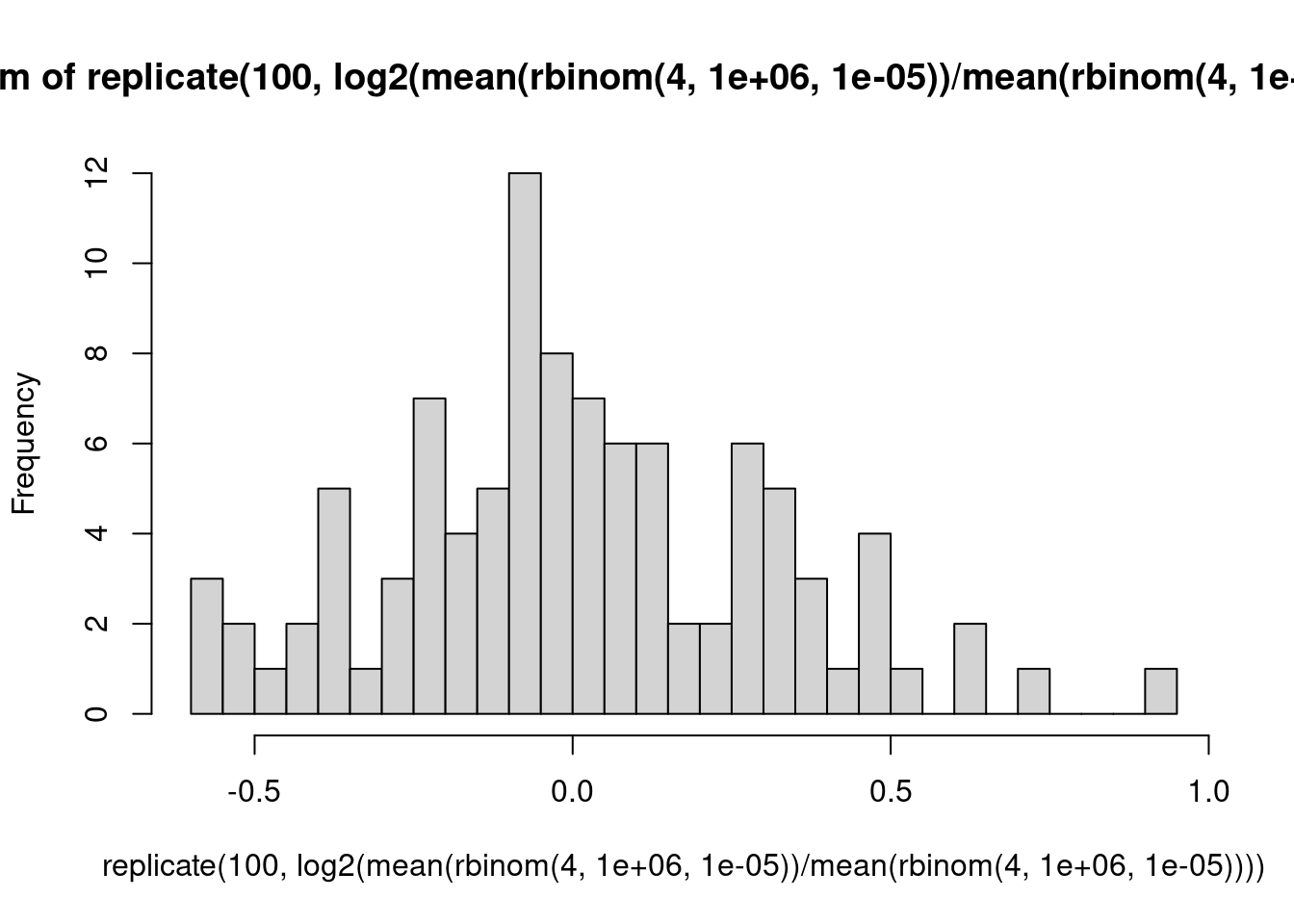
Ein Urnenmodell: Wir betrachten ein Gen mit eienr Expressionsstärke von 1000 CPM, d.h., ein Bruchteil von \(10^{-3}\) aller Reads kommt von diesem Gen. Für einen einzelnen Read beträgt die Wahrscheinlichkeit, dass er von diesem Read kommt, also \(10^-3\). Wir sequenzieren 1 Million Reads in der Probe. Wie viele werden auf dieses Gen gemappt? Wir simulieren dies 8 mal:
rbinom( 8, 1e6, 1e-3 )
[1] 1017 1017 986 979 992 1023 1000 1021
Was wäre, wenn wir über die ersten 4 und die zweiten4 Proben jeweils mitteln würden und das Verhältnis der Mittelwerte berechnen?
Folgerung: Bei schwach exprimierten Genen muss man auch unter der Nullhypothese deutliche Abweichungen des LFC von der Mitte (0) erwarten. Deshalb hat der MA-Plot links mehr “Dicke”, und deshalb braucht es dort auch sehr viel stärkere Effekte damit DESeq statistische Signifikanz meldet:
plotMA( dds )
Starke und schwache Effekte
Können wir dann Effektstärken (y-Werte im MA-Plot) überhaupt zwischen verschiedenen Expressionsstärken vergleichen? Nein, denn links im MA-Plot tendieren LFCs dazu, “übertrieben” zu sein.
DESeq2 hat eine Funktion, um dies zu kompensieren. Sie “schrumpft” die LFC-Werte zur 0 hin, wenn sie unsicher sind.
lfcShrink( dds, coef=2 ) -> res2
using 'apeglm' for LFC shrinkage. If used in published research, please cite:
Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
sequence count data: removing the noise and preserving large differences.
Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895